
EU Drug-Device Combination Products: Process Map
The EU regulatory landscape for drug-device combination products is evolving rapidly. With the new variation regulation now in place and the anticipated variation categorisation guideline expected in Q2 2025, another crucial piece of the puzzle is falling into place.
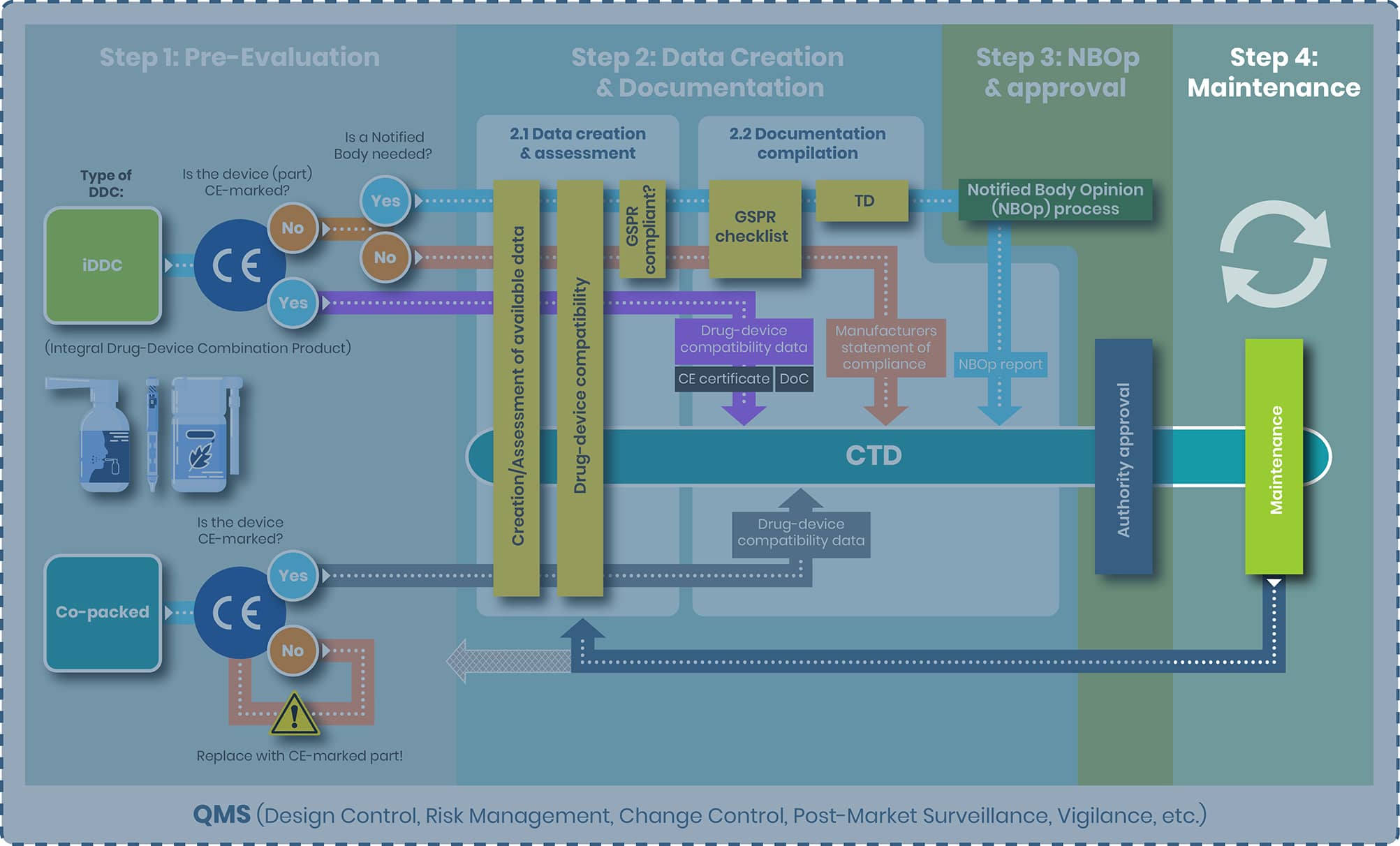
At regenold GmbH, we've been at the forefront of these changes, guiding our clients through the complex approval journey for combination products. To illustrate the procedure from start to finish, we have created a DDC process map outlining four distinct steps:
- Step 1: Pre-Evaluation
- Step 2: Data Creation & Documentation
- Step 3: NBOp & Approval
- Step 4: Maintenance
Step 1: Pre-Evaluation
Defining the Right Regulatory Path: Classifying Your Drug-Device Combination Product
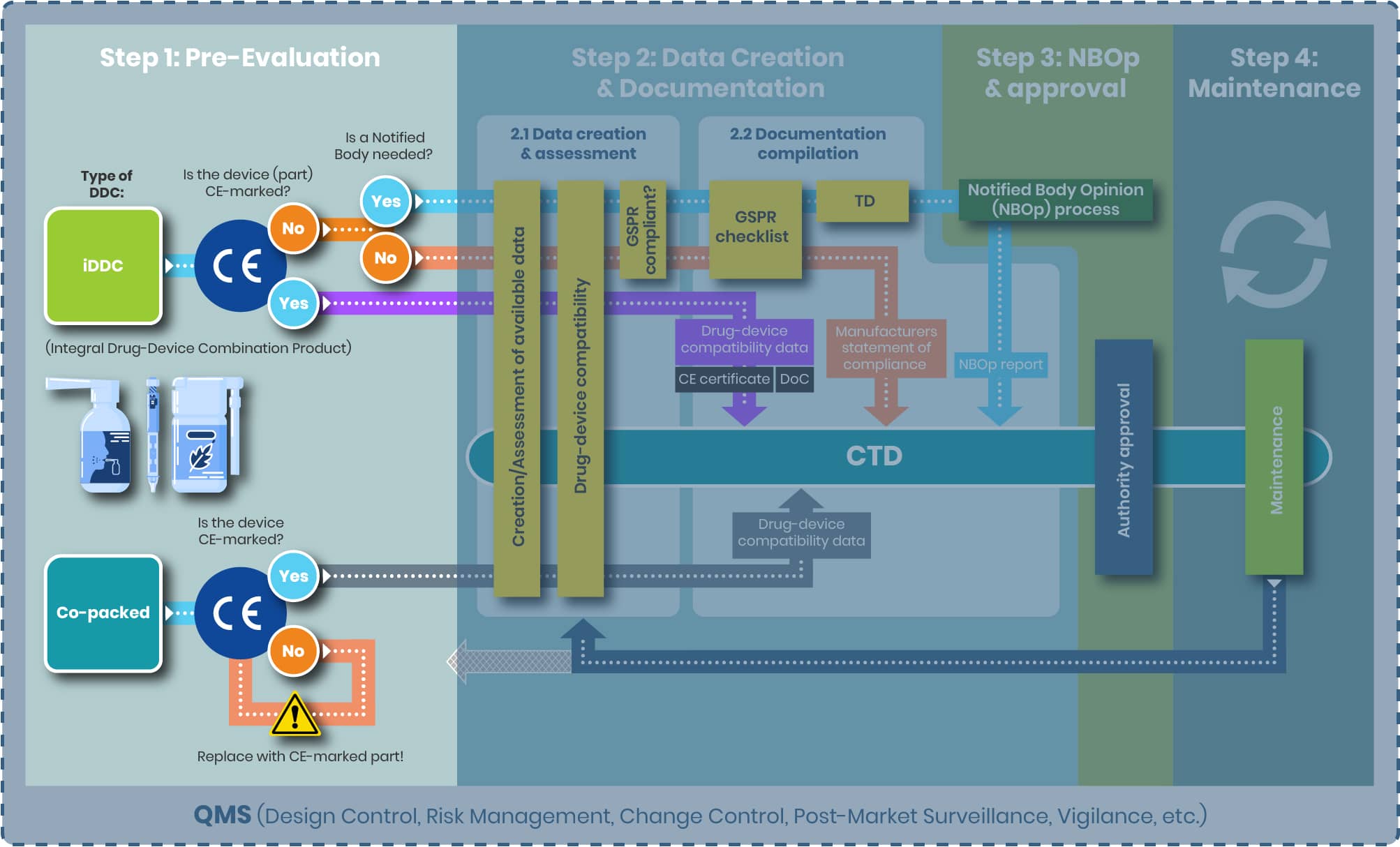
The first and most crucial step in the regulatory pathway is determining the correct classification of your product—whether it qualifies as an integral drug-device combination (iDDC), a co-packaged product, or both. Ideally, this decision should be made early in development and double-checked again for accuracy, as it defines the applicable regulatory route: either the EU Medical Device Regulation (MDR 2017/745) or Directive 2001/83/EC for medicinal products.
In the section that follows, we’ll explore key aspects of this classification, including:
- Differentiating between iDDCs and co-packaged products
- Verifying the need for a CE mark
- Understanding the role of Notified Bodies (NBs) in the approval process
These foundational elements are essential for a smooth and compliant development pathway.
iDDC or Co-Packaged: Determining the Right Classification
The EU regulatory framework for drug-device combination products follows a hierarchical approach, making early classification essential for compliance. Below are key distinctions:
- Integral Drug-Device Combinations (iDDC): These are single, combined products—such as pre-filled syringes—where the drug and device are physically integrated and not reusable. Governed primarily by Directive 2001/83/EC, the device component must meet relevant MDR safety and performance requirements.
- Co-Packaged Products: These involve a drug and a device supplied together but as separate items (e.g., a vial of insulin with a reusable injection pen). Such products fall under both the medicinal product and medical device regulations.
Note:
- Pure medical devices (e.g., empty syringes, wound dressings) are not covered here.
- Medical devices with an ancillary medicinal substance (e.g., a heparin-coated catheter) are also outside the scope of this section, though they represent a core area of our expertise.
Borderline cases can present challenges. The MDCG 2022-5 guidance provides clarity by outlining criteria related to physical integration, exclusive use with a specific drug, and reusability.
To support your regulatory strategy, the following decision tree offers step-by-step guidance in identifying the correct classification and regulatory pathway for your combination product.
CE Mark and Notified Body Involvement: Key Steps in the Regulatory Assessment
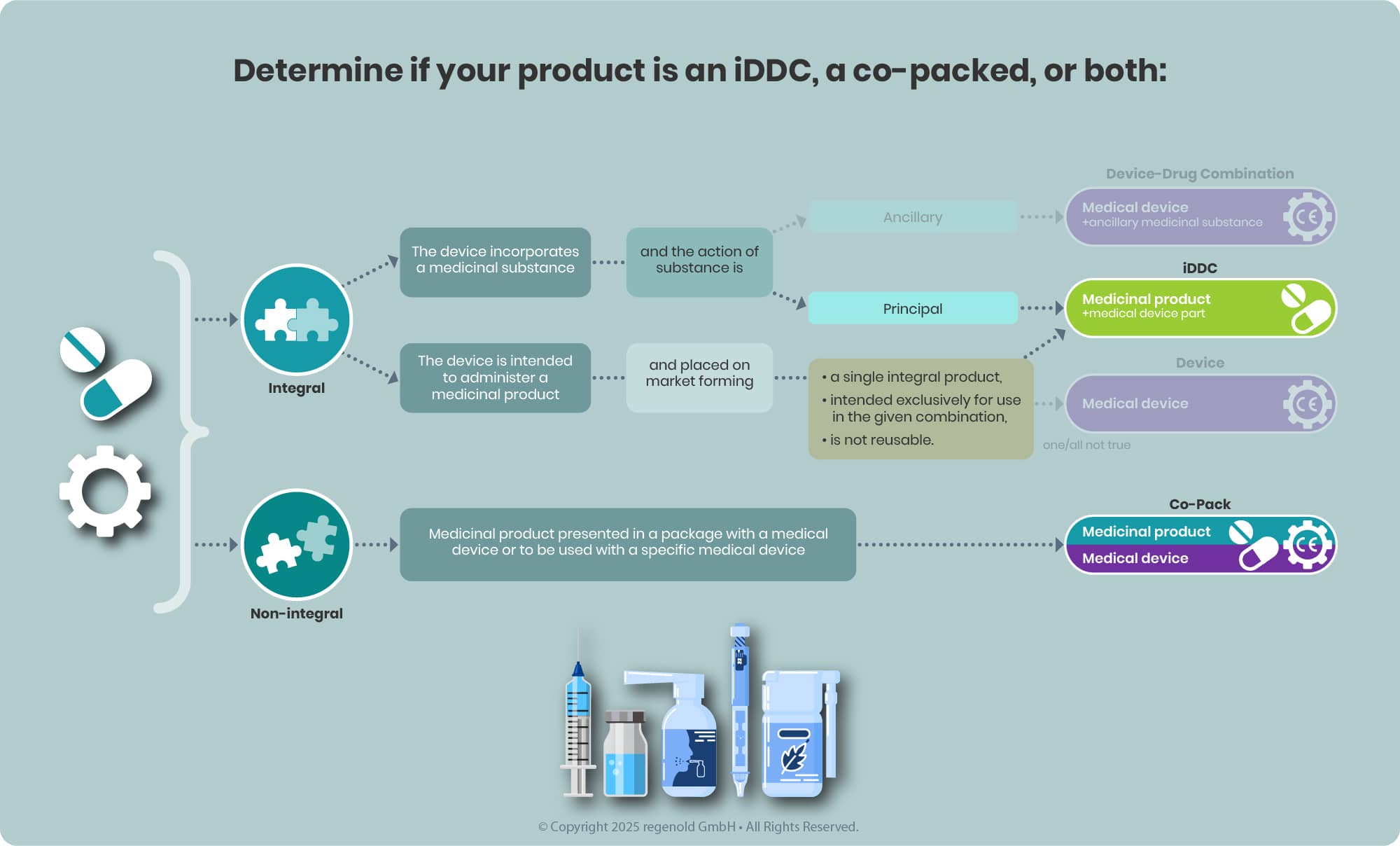
Once the classification of your combination product is determined, the next step is verifying the regulatory status of the medical device component—particularly its CE marking and the involvement of a Notified Body (NB).
CE Mark Verification
Start by evaluating whether the device component already holds a valid CE mark under the Medical Device Regulation (MDR). It’s essential to ensure that its intended purpose aligns with the therapeutic or medical function of the combination product.
- CE-marked and compatible: If alignment is confirmed, proceed confidently to the next step in the regulatory pathway.
- No CE mark or not compatible:
- For co-packaged products, identify a suitable alternative that meets MDR requirements.
- For iDDCs, classify the device component per MDR Annex VIII rules as it would be a standalone medical device.
Notified Body (NB) Involvement
The need for NB involvement depends on the device classification, which is determined based on factors such as the intended use, invasiveness, duration of contact, and the potential risk to users or patients—defined in detail in MDR Annex VIII.
- Class I devices (e.g., non-sterile syringes without measurement markings) typically do not require NB review.
- Higher-risk classes (Is, Im, IIa, IIb, III)—including sterile prefilled syringes or syringes with graduation—do require the involvement of a Notified Body seeking for Notified Body opinion (NBOp).
Understanding and fulfilling these requirements early in development ensures smoother regulatory progress and helps avoid costly delays.
Key Considerations for EU Regulatory Compliance of Drug-Device Combination Products
Successfully navigating the EU regulatory pathway for drug-device combination products requires a clear understanding of classification rules, applicable legislation, and the involvement of regulatory bodies.
Integrated vs. Co-Packaged: Choosing the Right Framework
- Integral Drug-Device Combinations (iDDCs) require a single marketing authorization under medicinal product legislation (Directive 2001/83/EC). However, the device component must comply to the applicable general safety & performance requirements (GSPRs) of Annex I MDR 2017/745.
- Co-packaged products, where the drug and device are supplied together but not physically integrated, are subject to dual regulatory frameworks, applying both medicinal and medical device requirements.
Device Classification: Why It Matters
The classification of the device component is critical and depends on:
- Intended use
- Sterility
- Functional features (e.g., presence of graduation markings)
For example, a prefilled syringe containing a medicinal product is considered an iDDC and regulated primarily as a medicinal product. However, its device component (the syringe) must still comply with MDR. If it is sterile or includes graduations, Notified Body (NB) involvement is mandatory.
This structured approach—guided by key EU documents—ensures regulatory compliance while providing clarity on product classification and oversight.
Essential References:
- MDR 2017/745
- EMA Guideline www.ema.europa.eu/en/quality-documentation-medicinal-products-when-used-medical-device-scientific-guideline
- EMA FAQ DDC www.ema.europa.eu/en/documents/regulatory-procedural-guideline/questions-answers-implementation-medical-devices-vitro-diagnostic-medical-devices-regulations-eu-2017-745-eu-2017-746_en.pdf
- MDCG 2021-24 www.health.ec.europa.eu/system/files/2021-10/mdcg_2021-24_en_0.pdf
- MDCG 2022-5 www.health.ec.europa.eu/document/download/b5a27717-229f-4d7a-97b1-e1c7d819e579_en?filename=mdcg_2022-5_en.pdf
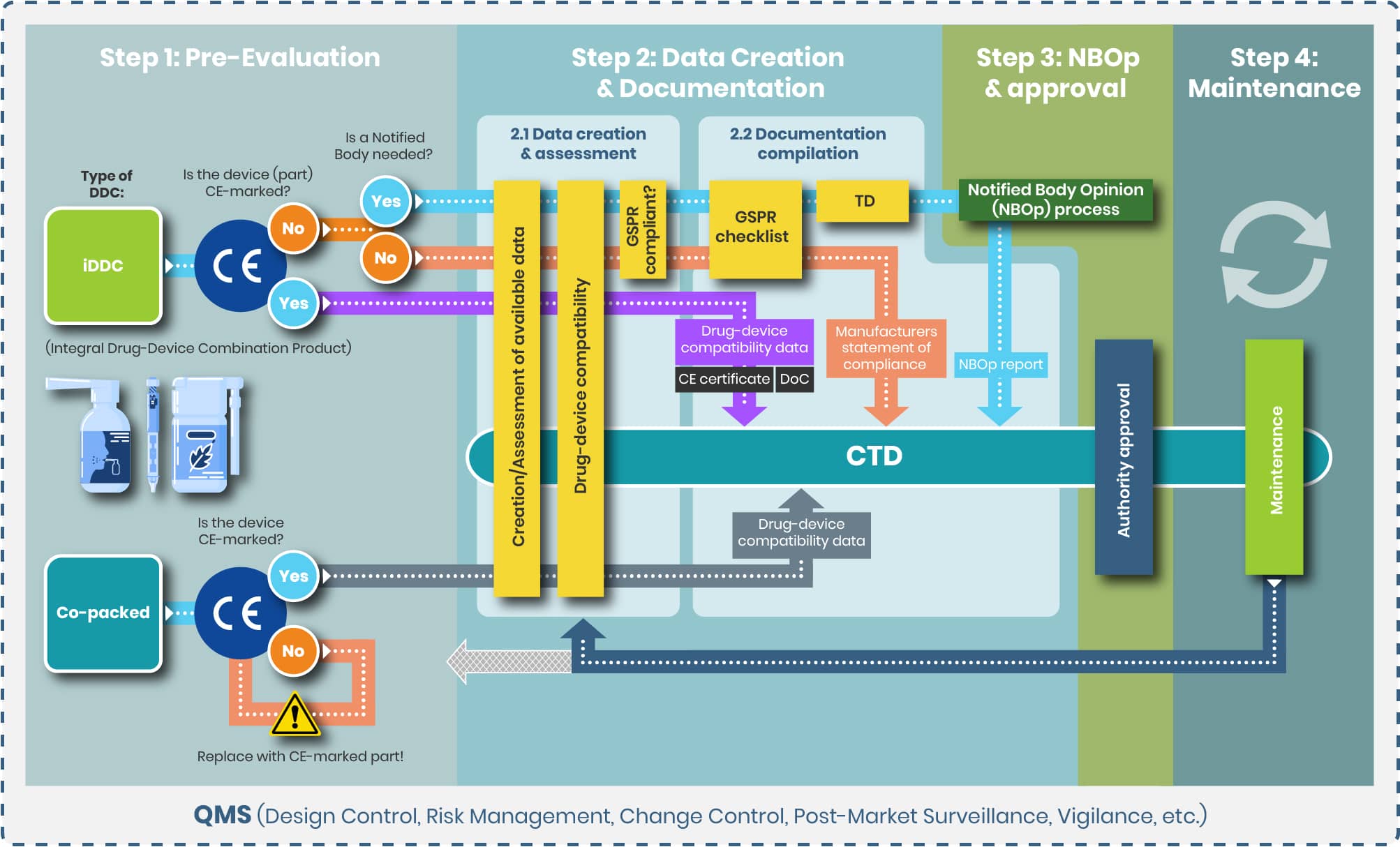
Step 2: Data Creation & Documentation
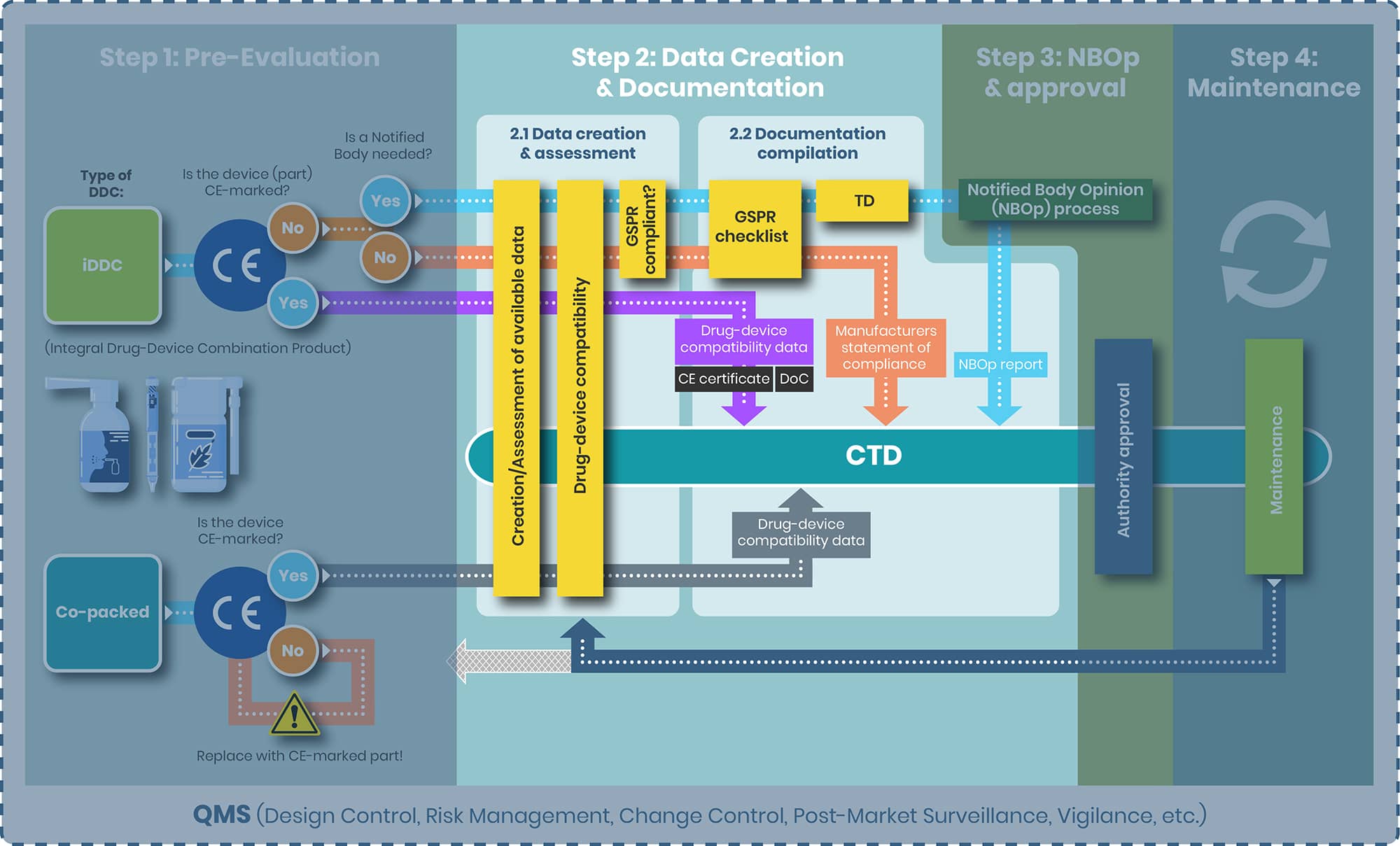
From top to bottom the tasks of step 2 and the substeps 2.1 and 2.2 are evaluated separately for the following products:
1. iDDC with and without NB
2. iDDC with CE-marked device component
3. Co-packed/ referenced
iDDC with and without NB
Step 2.1
Objective: For the iDDCs with and without a NB compliance to the applicable GSPRs, Annex I, MDR is to be demonstrated.
The GSPRs – the General Safety & Performance requirements are a set of requirements covering the whole range of medical devices. From a simple tongue plaster to MRT. For placing of a medical device on the EU market, compliance to the applicable GSPRs is to be demonstrated.
Annex I is divided into three main chapters: (details see below table)
- Chapter I general requirements
- Chapter II requirements regarding design and manufacture
- Chapter III requirements regarding the information supplied with the device
Covering requirements that ensure that device is:
- Safe for patients, users, and others
- Perform as intended
- Manufactured and designed to minimize risk
Each chapter does include several requirements, as outlined in the table below. Each of those requirements includes a subset of requirements resulting in total of approximately 200 requirements.
Table 1 GSPR overview
| CHAPTER I GENERAL REQUIREMENTS | CHAPTER II REQUIREMENTS REGARDING DESIGN AND MANUFACTURE | CHAPTER III REQUIREMENTS REGARDING THE INFORMATION SUPPLIED WITH THE DEVICE |
|---|---|---|
| 1 Safe; perform as intended | 10 Chemical, physical and biological properties | 23 Label and instructions for use |
| 2 Risk reduction, risk-benefit ratio | 11 Infection and microbial contamination | |
| 3 Risk Management System | 12 Devices incorporating a substance considered to be a medicinal product | |
| 4 Risk control measures | 13 Devices incorporating materials of biological origin | |
| 5 Eliminating & reducing risks related to use error | 14 Construction of devices and interaction with their environment | |
| 6 Lifetime | 15 Devices with a diagnostic or measuring function | |
| 7 Transport & Storage | 16 Protection against radiation | |
| 8 Minimising foreseeable risks, and any undesirable side-effects | 17 Electronic programmable systems | |
| 9 Devices without a medical purpose | 18 Active devices and devices connected to them | |
| 19 Particular requirements for active implantable devices | ||
| 20 Protection against mechanical and thermal risks | ||
| 21 Protection against the risks posed to the patient or user by devices supplying energy or substances | ||
| 22 Protection against the risks posed by medical devices intended by the manufacturer for use by lay persons |
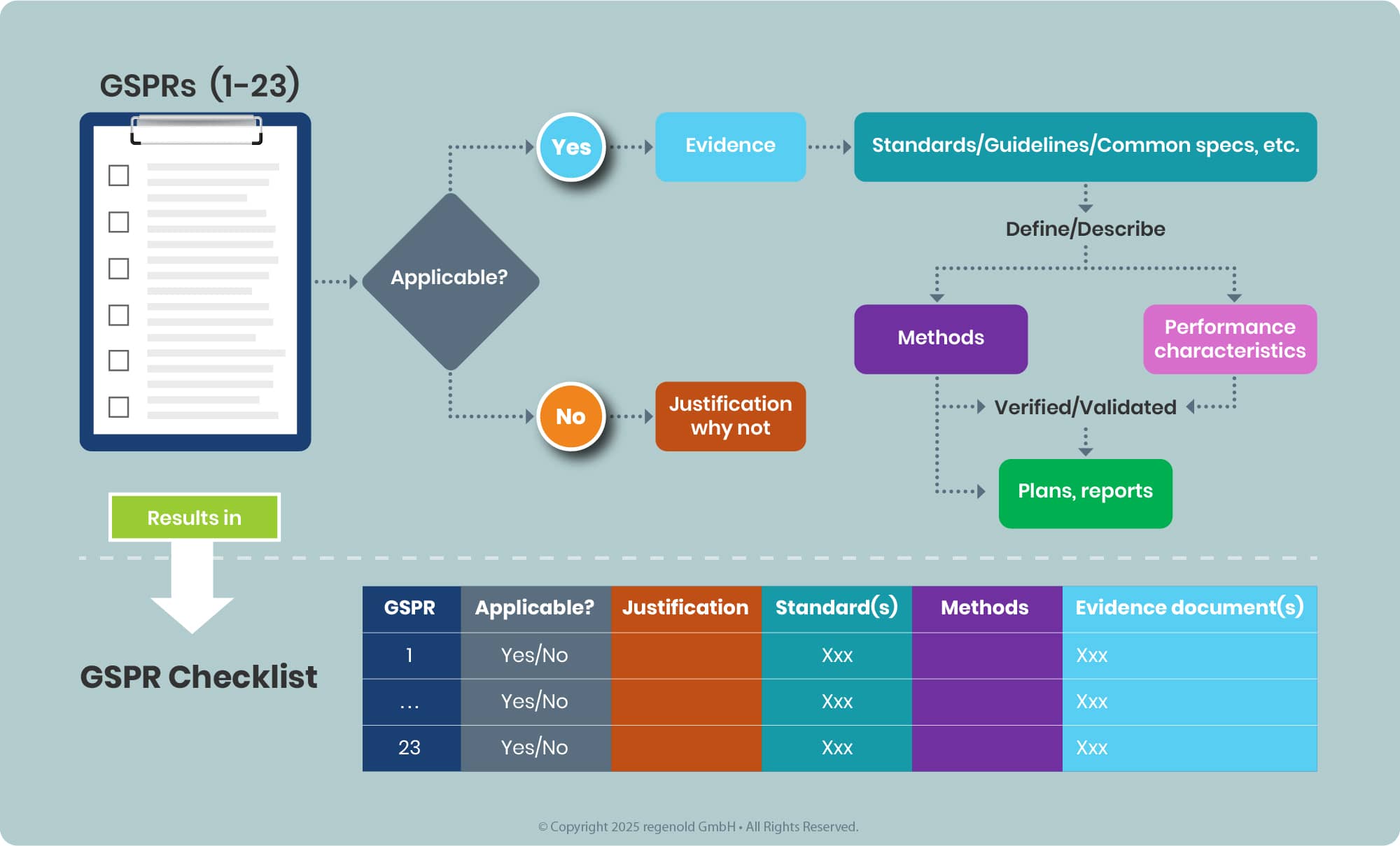
Documenting the tasks in a tabulated for this results in the so-called GSPR Checklist (see annex II MDR, 4), see example below.

In line with the latest EMA Q&A, the outcome of this assessment must be a conclusion of full compliance with all relevant GSPRs for the intended purpose of the device component used in the iDDC. Partial compliance conclusions or opinions that leave relevant GSPRs unaddressed are no longer acceptable, as medicines authorities do not reassess device GSPR compliance themselves. This applies both to Notified Body Opinions (NBOp) and, for eligible class I devices, to MAH GSPR compliance statements, which must clearly list the applicable GSPRs and confirm full conformity.
Step 2.2
While Annex I defines the WHAT (safety and performance requirements), Annex II specifies the HOW (documentation structure and data organization), detailing the technical documentation requirements necessary to demonstrate compliance to the applicable GSPRs.
That’s what some NBs expect being provided (and recommended as the review is facilitated by a familiar structure of the data).
Although not explicitly required by Article 117, Annex II not only provides a structure but also offers guidance regarding the content requirements for fulfilling Annex I.
For non‑sterile, non‑measuring, non‑reusable class I devices, the EMA Q&A allows the MAH to provide a MAH’s statement of compliance with the relevant GSPRs of MDR Annex I as an alternative, where a Declaration of Conformity is not available. This statement is only acceptable for that narrow subset of class I devices and must confirm full compliance with all relevant GSPRs, explicitly listing which GSPRs have been applied. For all other device classes, a valid EU certificate, NBOp, or manufacturer’s Declaration of Conformity is expected.
Table 2 Summary of Annex II MDR
| CH. | Description | Main Content | Key Points/Examples |
|---|---|---|---|
| 1 | Device description and specification, including variants and accessories | - General description of the device - Intended purpose and users - Risk class and justification - Principles of operation - Novel features - Accessories and variants - Reference to previous and similar generations |
- General description of the device - Intended purpose and users - Risk class and justification - Principles of operation - Novel features - Accessories and variants - Reference to previous and similar generations |
| 2 | Information supplied by the manufacturer | - Labels - Instructions for Use (IFU) - Other information provided with the device |
- All labeling and IFU as supplied to users |
| 3 | Design and manufacturing information | - Description of design and manufacturing processes - Manufacturing environment controls - Suppliers and subcontractors |
- Design drawings - Design Specifications - Manufacturing process details - List of critical suppliers |
| 4 | General safety and performance requirements | - Evidence of conformity with GSPRs (Annex I) - Declaration of conformity - List of applicable standards and guidelines |
- How the device meets safety/performance requirements - Applied harmonized standards |
| 5 | Risk management | - Benefit-risk analysis - Risk management process and results - Compliance with ISO 14971 |
Risk Management file Including Plan, report, analysis |
| 6 | Product verification and validation | - Pre-clinical and clinical data - Biocompatibility, electrical safety, software validation - Sterilization, stability, packaging - Additional info for specific cases (e.g., medicinal products, animal tissues, measuring function) |
Test reports/ test summaries, e.g. Design verification studies Biocompatibility plan & report, clinical evaluation, transport, packaging, sterilization… |
Why is that information about Annex II, I helpful?
Let us first take a closer look at the expectations regarding the data and information related to the medical device component within the dossier. These expectations are outlined in the EMA Guideline.
Some examples:
Module 3.2.
P.2 Pharmaceutical Development
This section of the dossier should summarise the information relevant to development of the specific medical device (part) integrated into the medicinal product, including the rationale for its selection in the specific sections of 3.2.P.2. A risk assessment summary for the medicinal product, aligned with relevant risk management principles in ICH Q9, should be presented.
P.3.5 Process validation and/or evaluation
Process validation for the integral medicinal product manufacturing process should be performed, as appropriate, in line with relevant European guidelines, including the assembly and sterilisation of the device (part) (if applicable) and any filling steps.
P.8 Stability
Stability studies for the integral medicinal product (or variant, where justified) should include the following tests/studies:
4. Functionality tests determined as stability-indicating CQAs for the medicinal product (refer to P.2.4).
The requirements defined by the EMA Guideline, the Annex II & I do correlate quite well, as shown with the examples below:
Table 3 Correlation between Annex II, I, & EMA guideline (excerpt)
| Annex II MDR sections | Annex I MDR | Dossier / CTD |
|---|---|---|
| 3 Summary of development 3a) information to allow the design stages applied to the device to be understood; |
1 “… devices shall be designed & manufactured…” See also, e.g. 7, 10 |
3.2.P.2 This section of the dossier should summarise the information relevant to development of the specific medical device (part) integrated into the medicinal product, |
| 3 Process verification & validation („ 3b (…) complete information and specifications, including the manufacturing processes and their validation“) | e.g. 1, 7, 10 | 3.2.P.3.5 |
| 6 Stability testing („ detailed information regarding test design, complete test or study protocols (…) for stability, including shelf life; “ | 6, 11.3, 11.4 | 3.2.P.8 |
Thus, this correlation might serve for various purposes, e.g.
- Guidance where to find data for the device component in the dossier (ideally) suitable demonstrating compliance to Annex I / II
- Guidance where to allocate device component data in the dossier
- Guidance where to allocate device component data in the TD
Even if it is considered as a single integral product, it will be unavoidable to divide the data in principle into three buckets, as these are used for different purposes.
- Part 1 and 2 for the Notified Body Opinion (NBOp) or the MAHs declaration of conformity.
- The MAHs declaration of conformity or NBOp plus bucket 3 and 2 for approval by the authorities.
Redundancies cannot be avoided, but can be reduced through a clever structure, which ultimately can also simplify data maintenance.
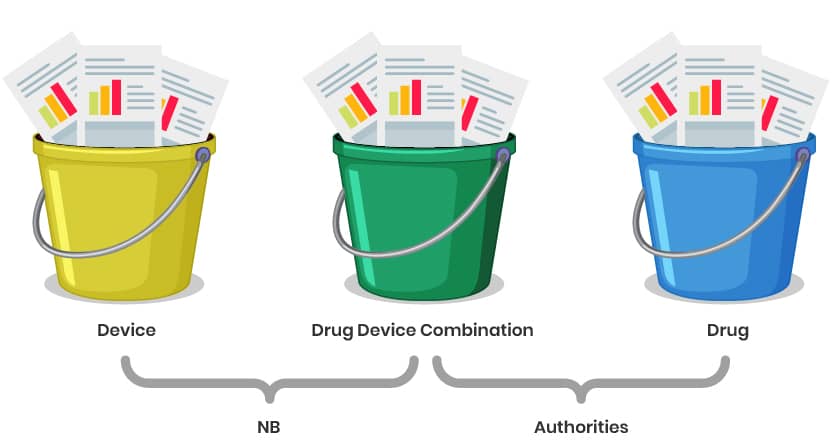
Explanation of the Structure:
Buckets 1 & 2: Focus on documentation for the Notified Body Opinion (NBOp) and MAHs declaration of conformity (e.g., GSPR checklist, risk management, technical specifications).
Parts 3 & 2: Combine the NBOp/ MAHs declaration of conformity with additional data (e.g., clinical, stability, or drug-specific information) for regulatory approval by authorities (e.g., EMA or national agencies).
Redundancy Management: Use cross-references and modular documentation to avoid duplication while ensuring all requirements for both the NBOp and marketing authorization are met. The correlation table between Annex II, I and the CTD might serve as a pointing document.
Data Maintenance: A well-organized structure ensures easier updates and compliance tracking over the product lifecycle.
iDDC with CE-marked device component
Step 2.1
Objective: Demonstrate compatibility and reconfirm the suitability of the device for use with the drug.
Data Assessment/Creation Steps:
- Reconfirm the scope of the CE-marked device for the combination product, including its intended use (indications, limitations), manufacturing processes, assembly requirements, and other relevant parameters.
- Evaluate existing data to determine its adequacy in addressing the requirements outlined in the EMA Guideline.
- Address potential gaps: While the CE mark provides a presumption of conformity with relevant requirements, additional data may still be necessary to fully demonstrate compliance with the General Safety and Performance Requirements (GSPRs) for the particular combination.
Step 2.2
Objective: Drug-device compatibility data
Documentation Compilation:
- Include a valid CE certificate or Declaration of Conformity (DoC) for the device to demonstrate compliance with applicable General Safety and Performance Requirements (GSPRs).
- Embed evidence documentation (e.g., supplier data, internal test reports, risk assessments) in the dossier to confirm compatibility between the device and medicinal product. This should address interaction risks, usability, and performance throughout the product’s lifecycle.
- Structure the data in line with CTD requirements provided in the EMA guideline
Key Considerations:
- Supplier Data: If relying on supplier-provided evidence (e.g., biocompatibility, sterilization validation), ensure it is traceable and referenced in the dossier.
- Own Data: Include in-house compatibility studies (e.g., leachables/extractables, dose accuracy) to address gaps not covered by the CE mark or supplier documentation.
- Risk Assessment: Align with ICH Q9/ISO 14971 principles to justify compatibility and mitigate interaction risks.
Co-packed/referenced
Step 2.1
Target: Demonstration of compatibility of the device with the drug.
Data Assessment/Creation:
- Request and assess device supplier data, including the CE certificate, scope of certification, and additional documentation such as the Instructions for Use (IFU) and any other relevant technical or regulatory documents.
Avoid assuming device manufacturer obligations under MDR Article 16 by ensuring:
- The device’s intended use remains unchanged.
- No modifications are made to the device that could void its CE mark - Follow EMA Guideline requirements to ensure alignment with regulatory expectations for a co packed/ referenced product
Step 2.2
Target: Drug-device compatibility data.
Documentation Compilation:
- Include a valid CE certificate or Declaration of Conformity (DoC) for the device to confirm compliance with applicable regulations.
- Embed evidence documentation (e.g., supplier-provided data, internal test reports, risk assessments) within the dossier to substantiate compatibility and safety of the device-drug combination. Ensure all data is traceable and cross-referenced in alignment with CTD structure (e.g., Module 3.2.P.7 for container closure system details).
Key Considerations
1. Regulatory Compliance and CE Marking
- CE Mark Validity: Ensure the co-packaged device has a valid CE mark under MDR (or MDD during transition periods). Verify the device’s intended use aligns with its original certification when paired with the medicinal product
- MDR Transition Challenges: If the device is transitioning from MDD to MDR, confirm the manufacturer’s compliance timeline to avoid market disruptions
- Unique Device Identification (UDI): Include the device’s UDI in documentation (if applicable) and ensure traceability
2. Documentation and Dossier Integration
- EMA Guidelines: Follow EMA’s quality documentation requirements for co-packaged products, focusing on:
- Container Closure System (Module 3.2.P.7): Detail device specifications, sterility, and compatibility with the drug
- Pharmaceutical Development (Module 3.2.P.2): Describe how the device impacts drug safety, efficacy, and usability (e.g., dosing accuracy)
- Stability Data: Provide in-use stability data if the device affects drug integrity (e.g., sterility maintenance) - Supplier Agreements: Secure contracts ensuring the device manufacturer remains responsible for MDR compliance (e.g., post-market surveillance)
Summary
Depending of the product, the following should be available:
| iDDC with NB | iDDC without NB | iDDC with CE marked device | Co-packed/ referenced | |
|---|---|---|---|---|
| Deliverable | GSPR Checklist Technical documentation acc. Annex II CTD |
GSPR Checklist Technical documentation acc. Annex II MAH’s statement of compliance with the relevant GSPRs of the MDR Annex I (only for class I devices) CTD |
GSPR Checklist (part of CE certificate/ Declaration of conformity for the device component, in a best case scenario) Suitability & compatibility data for the device component CE certificate/ Declaration of conformity for the device component CTD |
Suitability & compatibility data for the device component CE certificate/ Declaration of conformity for the device component CTD |
| Next step | NBOp | Authority approval | ||
Step 3: NBOp Process and Approval
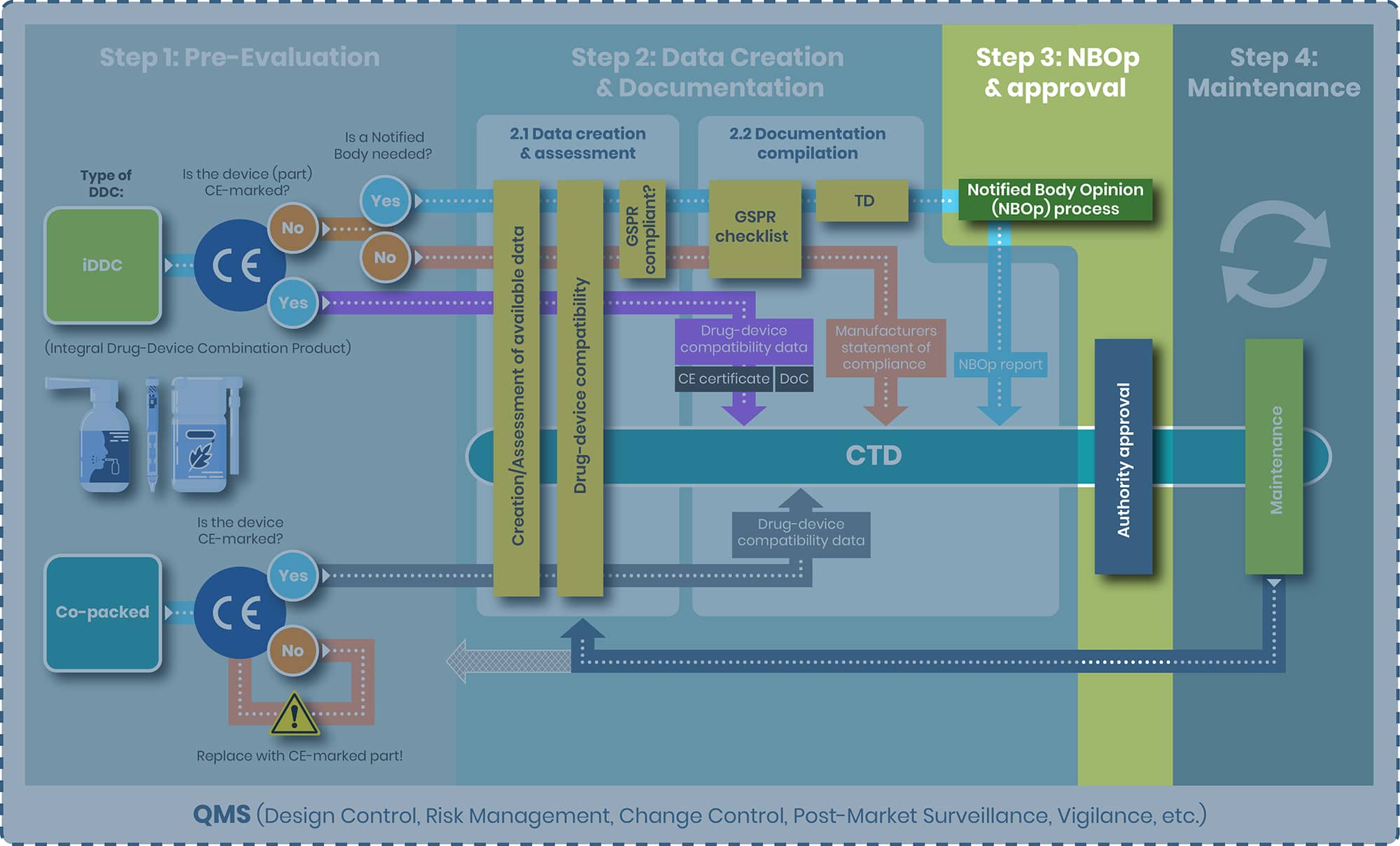
NBOp Process
For the iDDC requiring an Notified Body, Art 117 MDR mandates that manufacturer of the drug product (Marketing Authorization holder – MAH) must obtain a Notified Body Opinion (NBOp) on the device component before submitting their Marketing Authorization Application (MAA) to the competent authority, such as the European Medicines Agency (EMA). The NBOp process is not a certification or CE marking procedure; rather, the Notified Body conducts an independent assessment of the provided data demonstrating compliance to the applicable GSPRs using processes similar to those in a CE mark/conformity assessment. After this review, the Notified Body issues an Notified Body Opinion report, which must be included in the MAA.
The NBOp process under Article 117 does not assign ongoing surveillance responsibilities to the Notified Body for the device component. Therefore, the Notified Body involved in the initial NBOp may be different from the one involved in a subsequent NBOp, for example, if changes to the device require a new assessment.
However, the EMA Q&A now makes clear that the NBOp must confirm full compliance with the relevant GSPRs for the device component as used in the specific iDDC, including its intended purpose, user population and use setting. Opinions that only conclude on partial GSPR compliance or explicitly exclude relevant GSPRs are not acceptable to the medicines authorities and will need to be updated before an MA opinion can be issued.
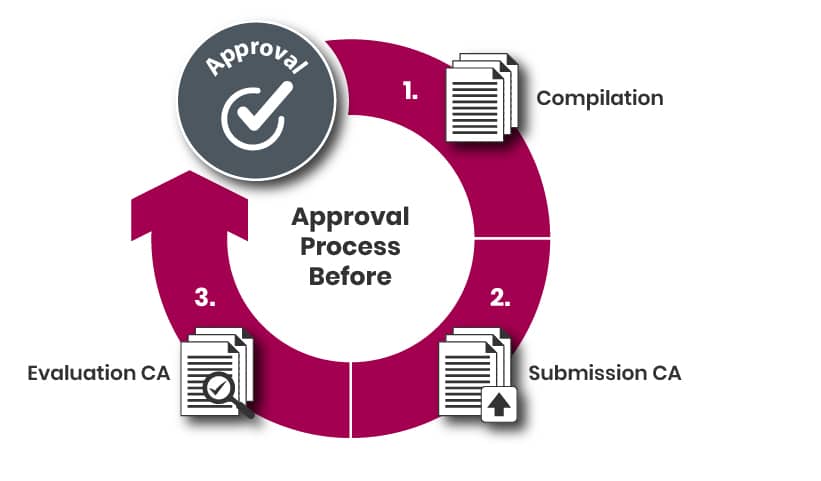
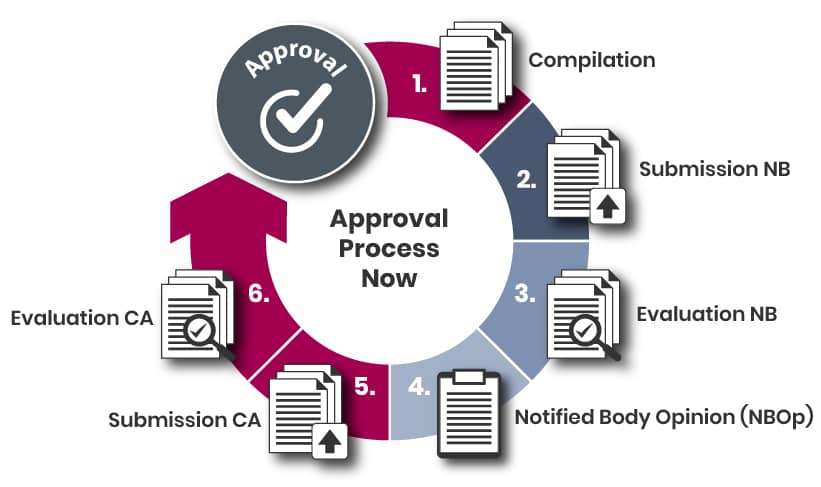
The steps leading up to the submission may include the following:
a) Identifying a suitable Notified Body;
b) Submitting an application to the selected Notified Body;
c) Receiving and approving a quotation;
d) Planning the submission process.
Identification of a NB (if not done yet)
The identification and selection of the Notified Body is the responsibility of the Marketing Authorization Holder (MAH). You can find a complete and regularly updated list of all Notified Bodies in the EU on the NANDO (New Approach Notified and Designated Organisations) website maintained by the European Commission. When choosing a Notified Body, factors such as the scope of designation, language, and other relevant criteria should be considered. Art 117 is not in scope of each notified body – early engagement is highly advisable.
It is possible scheduling structured dialogs with the preferred NBs prior to the application. Those discussions might be useful aligning on expectations, provide details on the product, etc. Per MDR, the NBs are not allowed to provide consultation, only discussions.
NBOp Application
Each Notified Body does have are more or less similar application process – completion of forms by the MAH or representative (either available on the website or upon request) followed by an offer related to the requested services.
Once having signed-off that quotation, the details relating to the submission can be discussed and scheduled.
Prior proceeding with the next step, it is highly recommended engaging with your NB early and align on expectations, if not yet done, like submission format, timelines, etc.
Submission & Evaluation
The specific details on what data to provide and how to structure it are outlined in Step 2 ("Data Creation & Documentation").
The Notified Body (NB) conducts its review and may issue questions or requests for clarification. The applicant is required to respond to these queries and provide any additional evidence as necessary. If, after three rounds of questions, unresolved gaps remain, the procedure is concluded with a negative outcome, and a negative NBOp report is issued.
Conversely, if all questions are satisfactorily addressed, the NB issues a positive NBOp report to the applicant.
Some notified bodies offer accelerated or dedicated review services, which can positively impact project timelines by expediting the assessment process. However, it is important to note that these services typically incur higher costs compared to the standard review procedure.
Timelines (might vary due due complexity/ ressources etc.):
- From identification to submission: 1-3m
- Submission to NBOp report: 3-6m
From a timesaving point of view, it seems logical to initiate the NBOp process with the submission to the authority. But, it poses a certain risk either not having completed the NBOp process in time or having a negative outcome.
For the other combination product types:
- iDDC without need for a NBOp
- iDDC with an already CE marked device
- Co-packed/ referenced (presuming the device is already CE marked)
There’s no need for a an additional review by a Notified body (presuming the device is and remains CE marked). The data relevant to the device component is to be provided with the submission to the authorities, see step 2 for further details about that data.
When and Why?
The requirements defined in Art. 117 and the EMA Guideline applies/ will apply to any drug-device combination product on the EU market considering the following timelines:
- Products approved prior to the date of application (DoA) of the MDR (26 May 2021) are not re‑opened solely because of Article 117. However, as soon as there is a major change to the device part that may significantly affect the safety, performance or intended use of the device, or when a new device/device part is added or replaces the existing one, a new or updated EU Declaration of Conformity, EU certificate or NBOp/MAH GSPR statement in line with MDR and Article 117 must be submitted within the appropriate post‑authorisation procedure.
- New marketing authorisations submitted on or after the MDR DoA must comply with Article 117 at the time of MAA submission, including provision of the relevant NBOp, certificate, DoC or MAH statement as applicable (see MDR, EMA Guideline & FAQ).
The fact that this approach is already being implemented is evident in the relevant sections of both the variation application form and the marketing authorisation application form. For example, in the variation application form (current version refer to https://esubmission.ema.europa.eu/eaf/index.html), applicants are required not only to indicate whether the submission concerns a change or a new device, but also to provide details regarding the type of combination product. In some cases, authorities already request the corresponding evidence as part of the submission process, while in other cases this is not yet required. This may be due to the current lack of a comprehensive legal framework. In step 4 of our series, we will further examine this topic, taking into account new and anticipated guidance, such as the Variation Categorisation Guideline.
Approval New Drug-device Combination Product
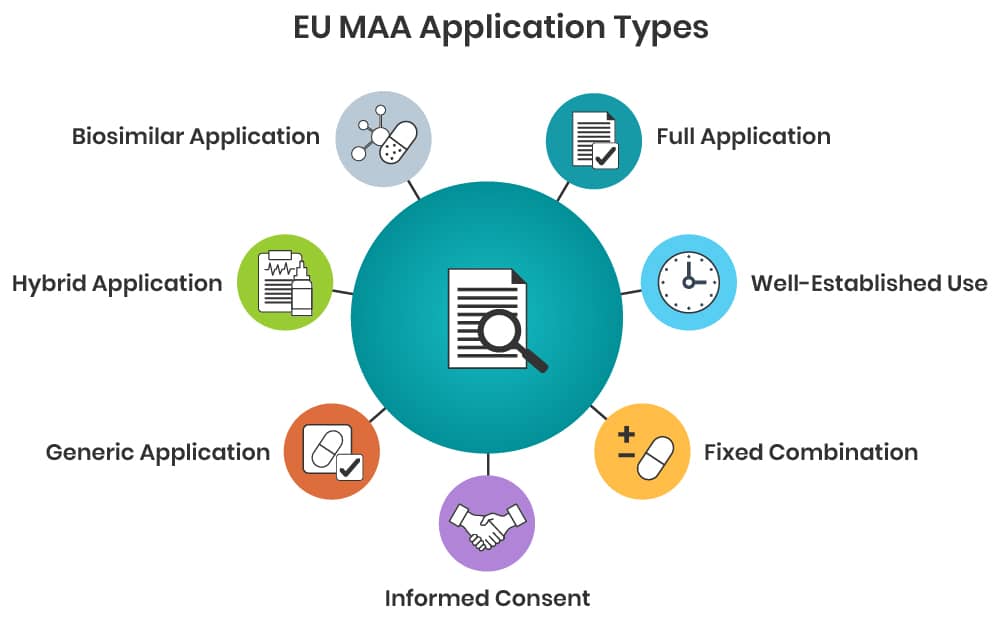
Legal basis of an EU MAA
Marketing Authorisation Applications (MAA) in the European Union (EU)/European Economic Area (EEA) are submitted in accordance with the relevant legal basis specified in Directive 2001/83/EC, as amended. The legal basis defines the regulatory requirements and documentation required as follows:
- Article 8.3 – “full” application supported by full clinical, non-clinical and quality documentation;
- Article 10(a) – “well established use” application supported by substantial published literature showing safe and efficacious use of the product for more than 10 years;
- Article 10(b) – “fixed combination” application whereby documentation usually expected to be linked to existing applications for the single active substances that are combined to achieve a therapeutic benefit;
- Article 10(c) – “informed consent” application whereby a MAH may grant another company full access to all pharmaceutical, clinical and non-clinical documentation for the purposes of applying for their own MAA for the same product;
- Article 10.1 – “generic” application whereby a MAH claims equivalence to the innovator product and reference to the innovator´s pharmaceutical, clinical and non-clinical documentation;
- Article 10.3 – “hybrid” application for products which do not fall into the definition of a generic medicinal product; supported by own data plus reference to the innovator’s pharmaceutical, clinical and non-clinical documentation;
- Article 10.4 – “biosimilar” application for biological medicines whereby a MAH claims equivalence to the innovator biological product and reference to the innovator’s documentation, as far as possible.
For a new drug-device combination product most likely Article 8.3, 10.3 or 10.4 becomes applicable. This should be evaluated in a product-specific regulatory strategy evaluation.
Regulatory Procedures
In general, within the EU/EEA there are four regulatory procedures for MAA, these are:
- Centralised Procedure (CP), whereby a single marketing authorisation granted in all EU/EAA countries at the same time - depending on the type of product the CP can be mandatory or optional;
- Mutual-Recognition Procedure (MRP), whereby a marketing authorisation granted in one EU/EEA Member State can be recognised in other EU countries;
- Decentralised Procedure (DP), whereby a medicine that has not yet been authorised in the EU can be simultaneously authorised in several EU/EEA Member States;
- National Procedure (NP), whereby a marketing authorisation granted in one EU/EEA Member State.
If the medicine is outside the mandatory scope of the centralised procedure and the medicine has not been authorised within the EU previously, the applicant/company can choose depending on their marketing approach which procedure to use.
Centralised Procedure (CP)
The Centralised procedure is a European Union/European Economic Area (EEA)-wide procedure for the authorisation of medicines, where there is a single application, a single evaluation and a single authorisation throughout the European Union. Only certain medicines such as products containing a new active substance to treat for example cancer, viral diseases or diabetes are eligible for the centralised procedure, details are available here.
EMA is responsible for evaluating the quality, safety and efficacy of marketing authorisation applications assessed through the Centralised Procedure, including the safety and performance of a medical device in relation to its use with a medicinal product.
Mutual-Recognition Procedure (MRP)/ Decentralised Procedure (DCP)
Both, the MRP and the DCP, are European procedure to apply for a marketing authorisation in the EU/EEA in more than one Member State. They are open for all applications which are outside the scope of the CP. The MRP must be chosen in case a marketing authorisation for the same medicinal product has already been granted by a Member State of the EU/EEA. Whereas the DCP is applicable if no marketing authorisation exists.
Both types of procedure are lead by the Reference Member State (RMS), who is responsible for preparing an Assessment Report (AR) which summarises the dossier presented by the applicant as well as characterises and critically evaluates the medicines quality, safety and efficacy. The RMS can either be chosen by the applicant (DCP) or is the MS of the already existing MA. All other countries are Concerned Member States (CMS), who review and evaluate the AR by the RMS. Each involved MS will issue a marketing authorisation based on the final outcome of the procedure.
In case consensus on the approvability of the MAA (or post approval variation) cannot be reached among the MS, the Coordination Group for Mutual-Recognition and Decentralised Procedure (CMDh) examines questions relating to these applications, facilitates the dialogue and helps finding an agreement.
Details information on the procedures and best practices guides are available on their website.
National Procedure (NP)
The national authorisation procedure is defined in the respective national law of each Member state. Information about the procedure and requirements can normally be found on the webpages of the national competent authorities. The list of CA can be found here.

The regulatory pathway for a Marketing Authorisation Application (MAA) for combination products requires early strategic planning to address complex compliance requirements and ensure timely approval. The process is shaped by the product’s classification, constituent components, and intended indication, necessitating a tailored approach from the outset.
Key Determinants of the Regulatory Roadmap
- Combination product classification (see step 1)
- Drug Substance or Indication
Orphan or pediatric indications require early engagement with the EMA’s Committee for Orphan Medicinal Products (COMP) or Pediatric Committee (PDCO). Orphan designation applications should precede MAA submission to leverage incentives, while Pediatric Investigation Plans (PIPs) must be agreed upon during development.
For novel indications or rare diseases, conditional MA or approvals under exceptional circumstances may apply, requiring post-authorization studies to confirm benefit-risk profiles.
Early Development Considerations and Strategic Elements
- Scientific Advice & Structured Dialogue: Proactive consultation with the authorities and/ or notified body is critical to align on non-clinical, clinical, and manufacturing requirements. This is particularly vital for innovative products deviating from standard guidelines
- Regulatory marketing planning: early considerations for market access strategies
- Exclusivity & patent research
Early definition of the regulatory roadmap, supported by multidisciplinary dialogue and adherence to evolving guidelines, is indispensable for navigating the complexity of combination product approvals
Re‑use of Existing Device Evidence in MRP/DCP and Line Extensions
For mutual recognition, decentralised procedures and line extensions of existing iDDCs, the need for updated device documentation depends on whether the device part has undergone significant change. If the same device is used without major changes to its design, performance or intended use, existing MDD‑based certificates and earlier device evidence may still be acceptable under the MDR transitional provisions. If a new device is introduced or a major device change has occurred, a new or updated NBOp, EU certificate, Declaration of Conformity or MAH GSPR statement in line with MDR and Article 117 will be expected as part of the procedure.
Further Links
- Authorisation of medicines | European Medicines Agency (EMA) (europa.eu)
- Obtaining an EU marketing authorisation, step-by-step | European Medicines Agency (EMA) (europa.eu)
- National competent authorities (human) | European Medicines Agency (EMA) (europa.eu)
- Medical devices | European Medicines Agency (EMA) (europa.eu)
- Quality documentation for medicinal products when used with a medical device - Scientific guideline | European Medicines Agency (EMA) (europa.eu)
- Heads of Medicines Agencies: Application for MA (hma.eu)
- Extensions of marketing authorisations: questions and answers | European Medicines Agency (EMA) (europa.eu)
Step 4: Maintenance
Same Same, But Different
Drug-device combination products-whether integral (iDDC) or non-integral/co-packaged-are fundamentally composed of three elements: the drug component, the device component, and the combination product as a whole.
Maintenance represents just one aspect of comprehensive lifecycle management, which encompasses the entire product journey from development through post-market activities (steps 1–4). Market feedback (such as complaints), evolving regulatory requirements, or supplier changes may trigger a formal change management process, typically involving the following steps:
- Definition of the change
- Identification and assessment of potential impacts
- Planning and execution of the change
- Final evaluation to ensure objectives are met
While these steps may be familiar from traditional pharmaceutical change management, the introduction of new regulations for combination products-particularly regarding the device component-brings additional terminology and topics. For example, it is now essential to determine whether a proposed change is considered substantial, and to document and justify this decision accordingly. Addressing these device-specific requirements is critical to ensuring regulatory compliance and maintaining product quality throughout the lifecycle.
The EMA Q&A now explicitly links such change assessments to Article 117 in the post‑approval phase. For all authorised iDDCs, regardless of whether they were originally approved before or after the MDR DoA, major changes to the device part that may significantly impact its safety, performance or intended use trigger the need for a new or updated EU Declaration of Conformity, EU certificate or NBOp/MAH GSPR statement to be included in the relevant variation or extension application. Contractual arrangements between MAH and device manufacturer should ensure timely notification and provision of updated device compliance evidence.
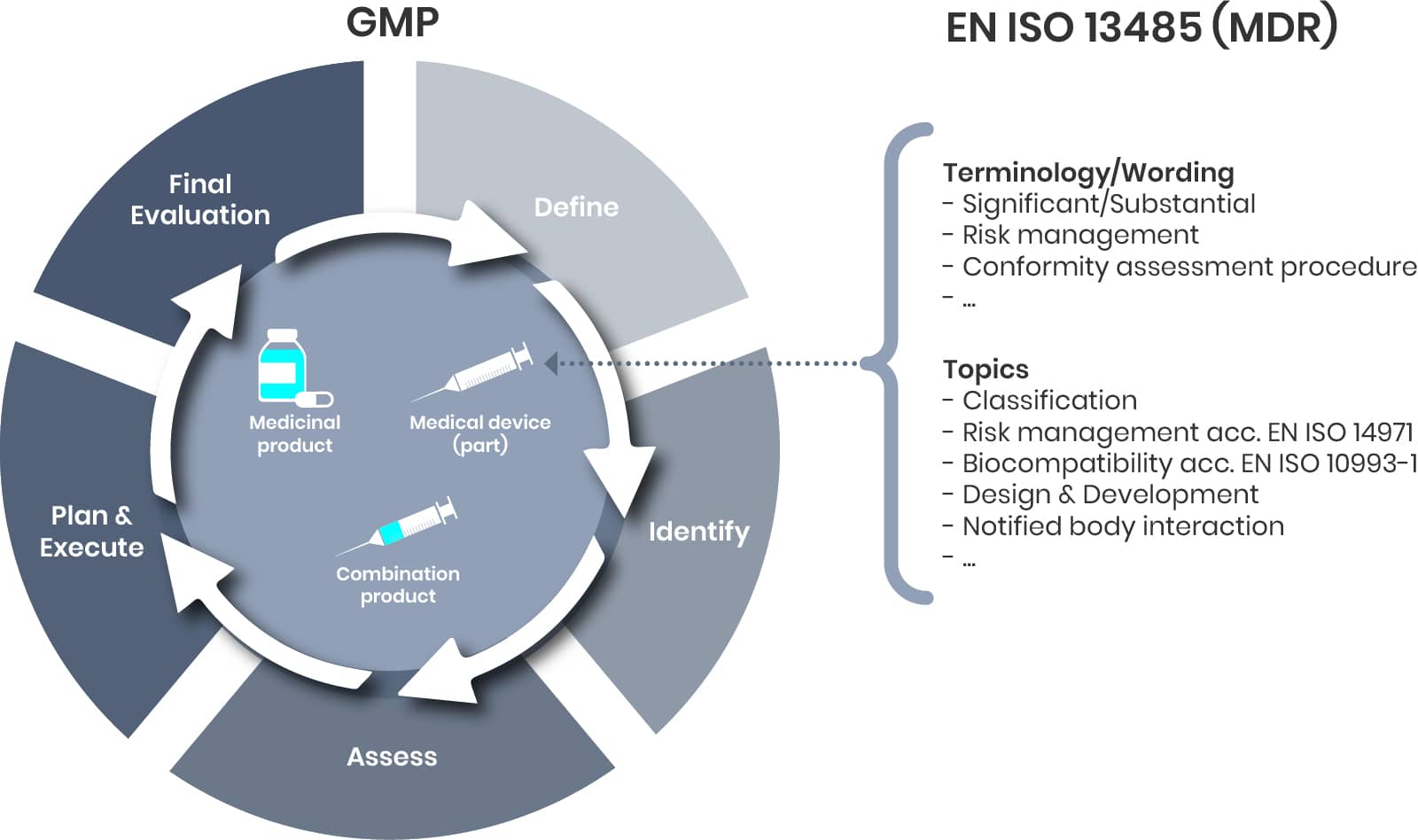
Since the date of application of the Medical Devices Regulation (MDR) in 2021, the regulatory landscape for combination products in the EU has evolved significantly, particularly in the area of lifecycle management. The forthcoming update to the variation categorisation guideline is expected to address existing gaps by introducing a more holistic approach to the regulation of combination products. This update will help ensure that both the medicinal and device components are consistently managed throughout the product lifecycle, reflecting the integrated nature of these products and aligning regulatory expectations across the EU.
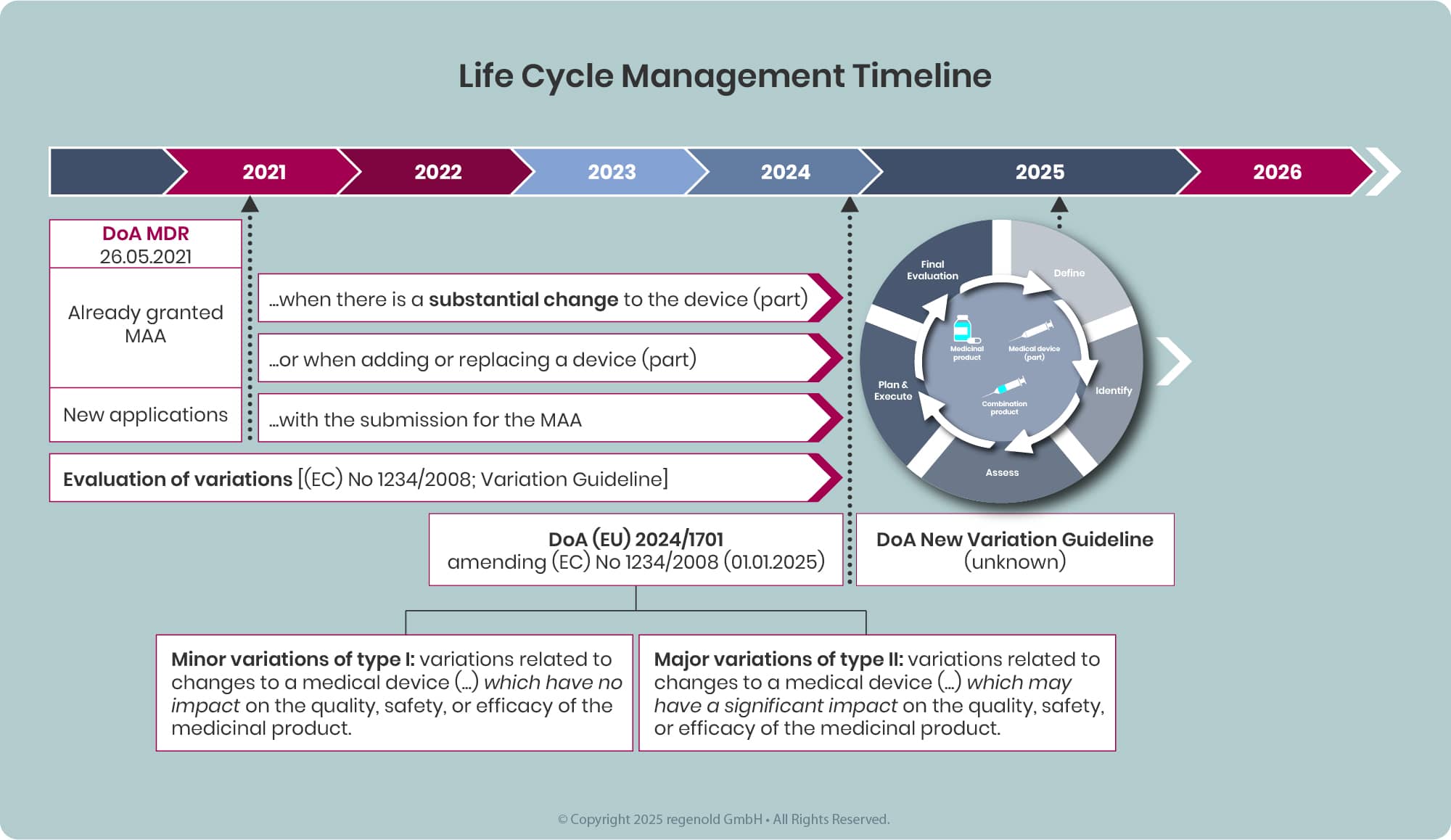
Any combination product available on the EU market, will sooner or later be affected by and required addressing the requirements defined in the MDR and EMA guidelines and additional legislation.
Changes to an Approved Drug-device Combination Product
During the lifecycle of an approved drug‑device combination product, the device component may need to be changed, replaced or newly added. The MAH is responsible for evaluating whether such changes could affect the delivery, quality, safety or efficacy of the combination product and for determining whether a variation or extension is required. In cases where changes may significantly affect device safety, performance or intended use, the EMA Q&A expects a new or updated EU Declaration of Conformity, EU certificate or NBOp/MAH GSPR statement to be submitted with the post‑authorisation application.
In addition, the MAH should determine the impact on the medicinal product quality, safety and efficacy profile and whether an update of the approved documentation (CTD dossier) is required.
As a general rule, a new or revised NBOp or MAH GSPR compliance statement is:
- required when a new device or device part is introduced with a line extension or variation;
- expected when major changes are introduced to an existing device, such as changes to design, key performance characteristics, intended purpose (including new patient population, different user such as home vs hospital, or significantly different instructions for use/usability);
- expected when changes to the medicinal product (for example a new formulation with markedly different viscosity) may impact device performance or safety.[1]
Depending on the type of change either a Variation or an Extension application becomes applicable.
Variation
Variations to the dossier should be submitted in accordance with Regulation (EC) No 1234/2008 (Variation Regulation), as amended, and associated variation guidelines. This also applies for changes related to a device (component).
Each variation to be categorized considering the impact of the change on the quality, safety or efficacy of a medicinal product, particularly where a change impacts any medicinal product Critical Quality Attributes (CQA) and/or any element of the overall medicinal product control strategy.
In general, the Variation Regulation defines variations as minor (Type IA, Type IB) or major (Type II). Details to the variation procedure, Q&A and BPG are provided on the EMA (here) and CHMP (here) webpages.
Extension
Certain changes to an approved marketing authorisation have to be considered to fundamentally alter the terms of this authorisation and therefore cannot be granted following a variation procedure. These changes are to be submitted as Extension application and are listed in annex I of the Variations Regulation.
The three main categories of 'changes requiring an extension of marketing authorisation' are:
- changes to the active substance;
- changes to the strength, pharmaceutical form and route of administration;
- other changes specific to veterinary medicinal products to be administered to food-producing animals or change or addition of target species.
Further guidance is provided by EMA, here and in the Guideline on the categorisation of new applications versus variation applications.
The assessment of changes for integral Drug-Device Combinations (iDDCs) and co-packed combination products is a complex and nuanced process that should not be underestimated. Each case requires careful, context-specific evaluation and often demands a fresh interpretation of the applicable regulatory framework. This is particularly relevant within the European Union, where the regulatory landscape is continuously evolving. The forthcoming Variation Regulation Guideline is expected to further clarify these requirements, underscoring the necessity for a thorough and case-by-case approach to change management in combination products. The following examples are intended to illustrate these complexities in greater detail and demonstrate why a diligent, individualized assessment is essential for ensuring regulatory compliance and patient safety.
iDDC - Example 1
Let’s assume the following. For an iDDC (e.g. prefilled syringe), the patient population is to be extended covering children. Beside evaluating this extension from a drug point of view it’s now defined evaluating that change from a device point of view.
And that’s a different story than the variation categorisation. According to the TEAM NB Guidance
“A change of the device component of the medicinal product is understood to be substantial or significant if it may affect the device conformity with the General Safety and Performance Requirements.“
Or as outlined in the EMA FAQ for DDC Rev. 5
“As a guiding approach, a new or revised notified body opinion or for class I (excluding Im and Is), MAH’s GSPRs compliance statement for the device part of an iDDC is:
1) required when a new device is introduced with a line extension or variation;
2) expected when major changes are introduced to an existing device, such as:
- change to its design;
- addition or replacement of an integral device (part)
- change to its performance characteristics;
- change to its intended purpose such as a different patient population and/or new user (e.g. home versus hospital setting) and/or new usability study, and/or significantly different instructions for use.
which may have a significant impact on the delivery or the quality, safety, or efficacy of the medicinal product.”
(For more details, please refer to the guidance documents)
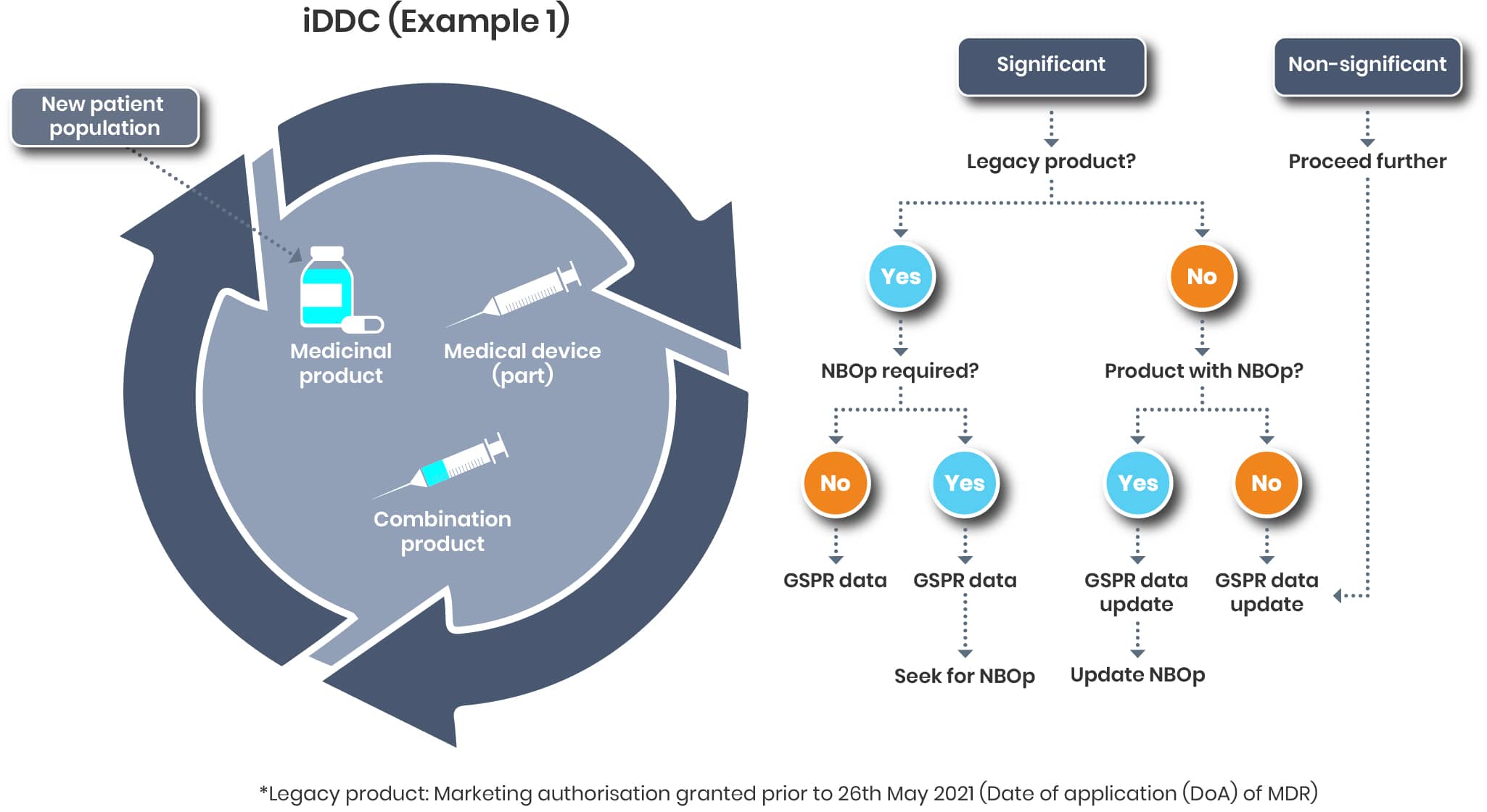
A general statement as to when a change is considered significant or not cannot be made. Each case must be assessed individually, taking into account the relevant regulations from both the medical device and pharmaceutical sectors.
Changing the patient population will most likely this result in a significant change (https://www.team-nb.org/wp-content/uploads/2025/02/Team-NBPosition-Paper-Art117SubChangeLifeCycleMngt-202012.pdf)
The further activities will then depend on the regulatory status of the product affected.
So, you might find yourself back in step 1 as it is not clear or you might start in step 2 of the process.
Example 2:
Let us consider a scenario where a material modification is required for a device component, such as the outer shell of an autoinjector. Although this outer shell typically does not come into direct contact with the drug substance—thereby often being classified as a minor variation—from a device perspective, it maintains direct contact with the patient or user. Consequently, this change could impact compliance with the applicable Good Safety and Performance Requirements (GSPRs) and should therefore be evaluated as a substantial change. This assessment may necessitate the initiation of a new Notified Body Opinion (NBO) or the pursuit of an updated NBO.
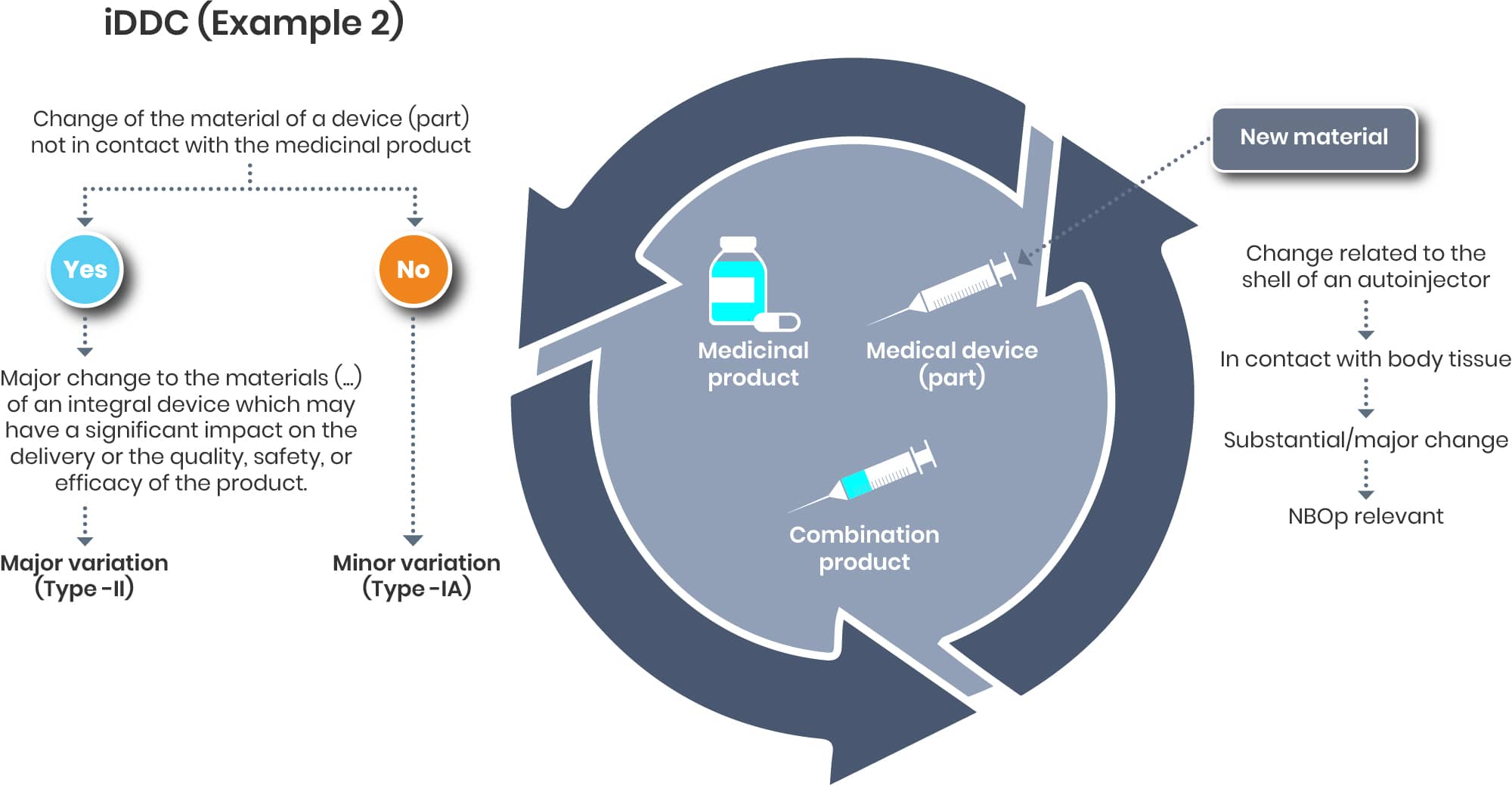
A change of the shell of the autoinjector might be considered as significant on the device and as a minor on the drug end.
If such cases might not end up as a major change as it could be argued that the change may have a significant impact on the delivery is not clear yet.
Examples for Co-packed Products
For a co-packed product, it’s a bit different. The pivotal question will be if the new patient population is covered by the certificate/declaration of conformity of the device or not.
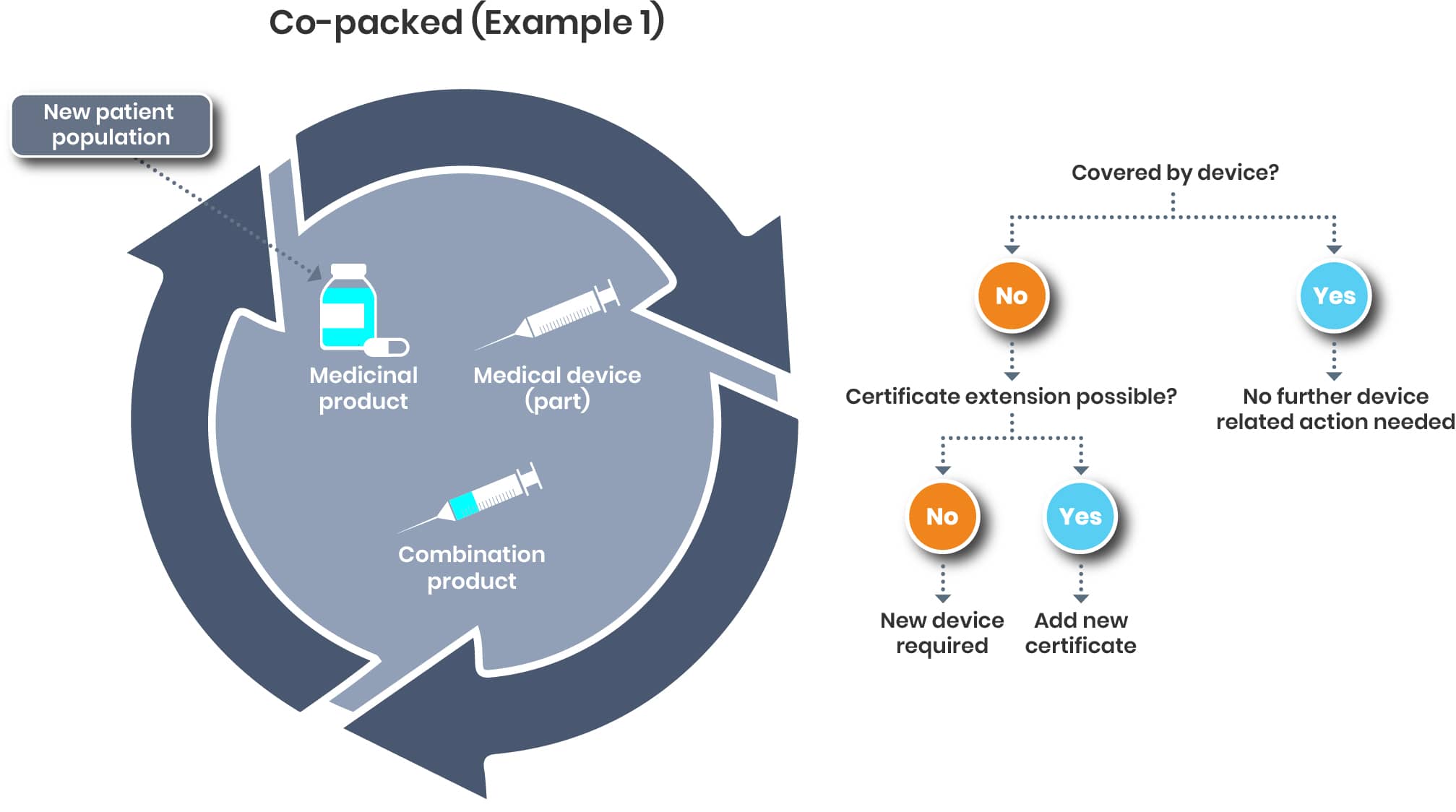
The regulation of drug-device combination products is evolving quickly and requires additional efforts.
Co-packed Example 2
In case of a material change of the co-packed device, the impact on the drug/ medicinal substance is also to be evaluated and the respective measures to be taken.
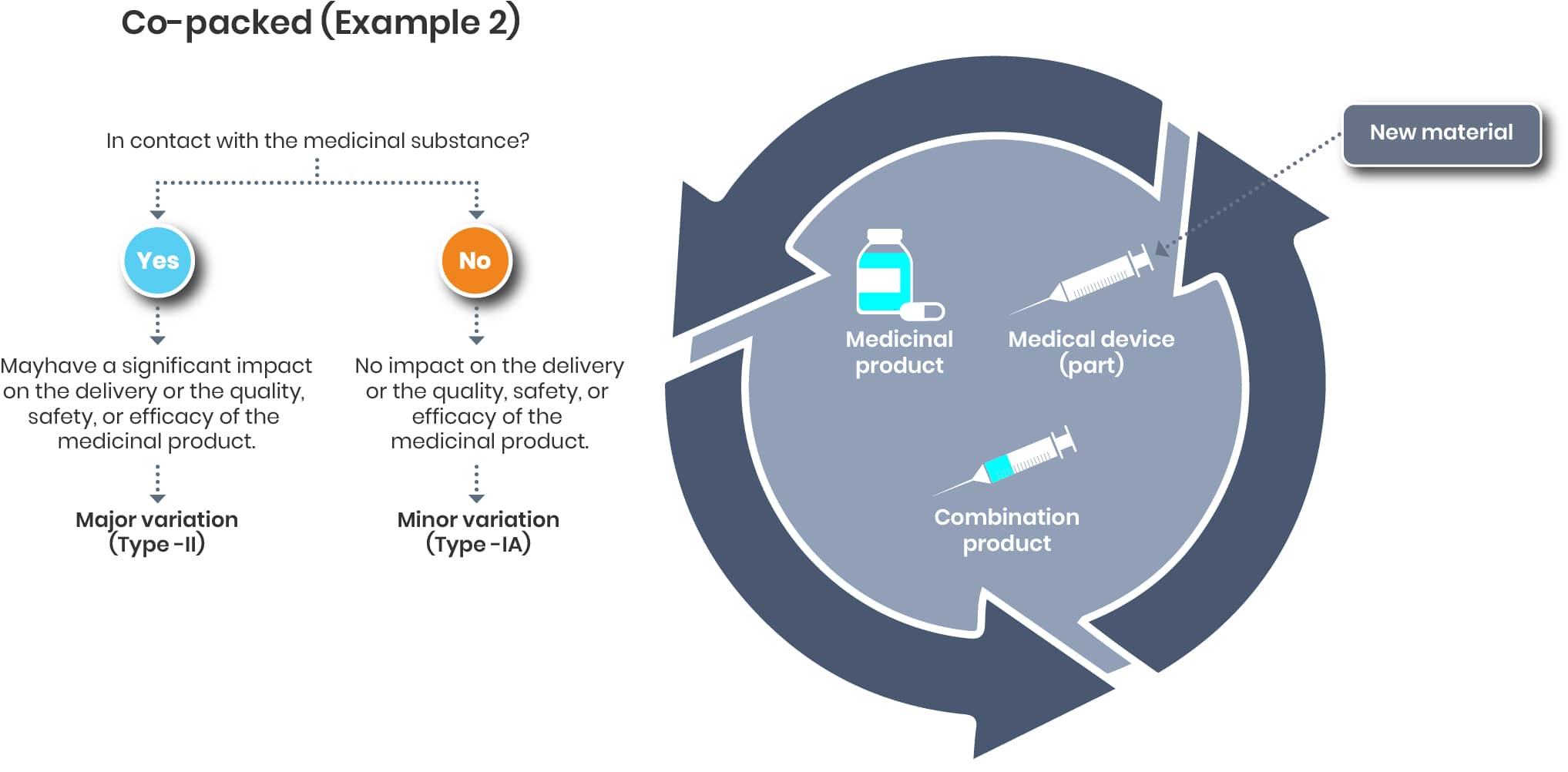
The evaluation on the device end is up to the Legal manufacturer of that device. If needed, the renewed certificate/ declaration of conformity should be added to the dossier once available.
Ongoing maintenance is a critical phase in the lifecycle management of drug-device combination products.
Effective lifecycle management for drug-device combination products in the European Union is both critical and highly complex. This complexity arises from the need to continuously ensure compliance with evolving regulatory requirements, not only for newly developed products but also for those already on the market. It is essential that existing products are regularly reviewed and reassessed, with appropriate actions taken based on up-to-date regulatory guidance and product-specific considerations.
We strongly recommend proactively evaluating your entire product portfolio to identify the presence of combination products. This includes determining the correct product classification—such as integral Drug-Device Combinations (iDDCs) or co-packed products—and clarifying whether involvement of a Notified Body is required. This foundational step (step 1 of the process) is crucial for establishing a compliant and efficient lifecycle management strategy.
Additionally, it is advisable to review the current data and documentation for each product. This enables the identification of any gaps or necessary updates, ensuring that upcoming variations can be managed in a timely and successful manner. Beyond regulatory compliance, this process also touches on a range of related topics, such as risk management, supply chain considerations, and patient safety, all of which must be carefully addressed as part of a comprehensive lifecycle approach.
Summary:
In a nutshell, the 4 steps are key to obtaining regulatory compliance of drug-device combination products.
Step 1: Pre-Evaluation
- Systematically review your product portfolio to identify all drug-device combination products, including both new and existing products.
- Classify each product accurately as either an integral drug-device combination (iDDC) or a co-packaged product, referencing the latest EU guidance (e.g., MDCG 2022-5).
- Verify if a Notified Body (NB) involvement is required, based on device classification, intended use, and risk class.
- Document the regulatory pathway for each product, ensuring clarity on whether MDR, Directive 2001/83/EC, or both apply.
Step 2: Data Creation & Documentation
- Assess and update technical documentation for each combination product, ensuring alignment with current regulatory requirements (including GSPRs for device components).
- Review and compile all supporting data (clinical, technical, quality) to identify any gaps or areas needing improvement.
- Ensure traceability and consistency between drug and device documentation, especially for integral products.
Step 3: Notified Body Opinion (NBOp) & Approval
- Engage with Notified Bodies early if required, especially for higher-risk device components.
- Prepare and submit robust NBOp dossiers or other regulatory submissions as needed, addressing all identified gaps.
- Monitor approval progress and be ready to respond to regulatory queries or requests for additional information.
Step 4: Maintenance
- Implement a continuous lifecycle management process for all combination products, including regular review of regulatory status and compliance.
- Monitor evolving EU regulations (such as the anticipated Variation Categorisation Guideline) and assess their impact on your product portfolio.
- Plan and execute timely variations to ensure ongoing compliance and uninterrupted market access.
- Regularly review and update product data and documentation, addressing new requirements or changes in product classification.
Learn More:
Roles and Responsibilities
Roles and Responsibilities under the MDR
Under the EU Medical Device Regulation (MDR), a “manufacturer” isn’t always the one physically making the product. Confused? You’re not alone. MDR changes the way roles and responsibilities are defined — and these distinctions have real compliance consequences.
Why This Matters
Knowing exactly who is responsible for what is essential for meeting regulatory requirements, avoiding delays, and ensuring product safety. This is especially true for integral and co-packaged drug-device combination products, where different parties may carry overlapping duties.
Key Economic Operators (MDR)
The MDR defines several “economic operators,” each with specific legal responsibilities:
Manufacturer
A person or company that manufactures, refurbishes, or has a device manufactured/refurbished and markets it under their name or trademark (MDR Art. 2(30)). Clear tasks defined in Art. 10.
- Legal manufacturer: Legally responsible for the device, even if a contract manufacturer (CMO) actually makes it.
- Comparable to the Marketing Authorisation Holder (MAH) for medicinal products.
- Often misunderstood: in pharma, “manufacturer” usually means the physical maker; in MDR terms, this is about legal responsibility, not production activity.
- Identified on packaging next to the manufacturer symbol.
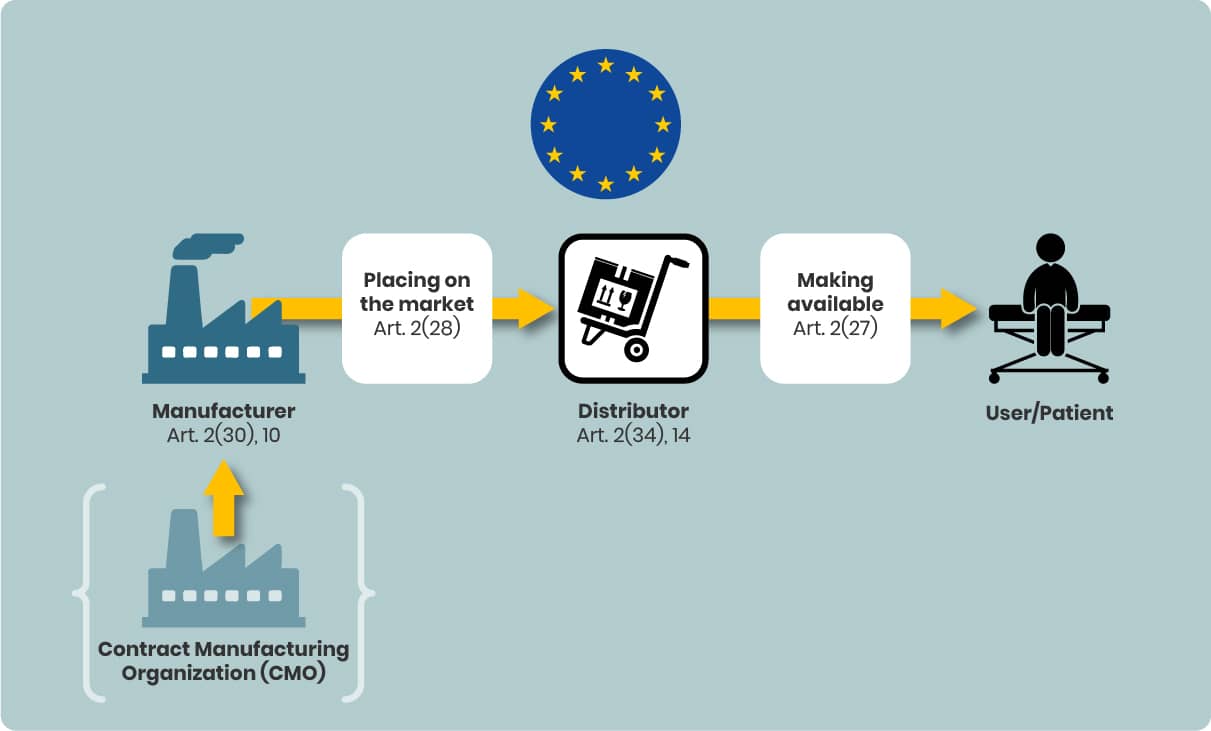
Bringing a Device to the EU Market: The Process
The MDR structures market entry into distinct steps, in order:
- Placing on the market – First making the device available in the EU (MDR Art. 2(28)).
- Making available on the market – Any commercial supply of the device (MDR Art. 2(27)).
- Putting into service – When the device is ready for first use by the final user for its intended purpose.
Takeaway:
Under the MDR, “who manufactures” and “who is responsible” are not always the same — but compliance obligations follow the legal definitions, not the production reality. Clearly defining these roles from the start prevents confusion, ensures regulatory compliance, and smooths market entry.
Considerations for iDDCs and Device Component Choice
For iDDCs, the overall legal responsibility lies with the Marketing Authorisation Holder (MAH) of the medicinal product.
Key clarifications:
- The legal manufacturer need not be the physical manufacturer of the device (part); manufacturing can be outsourced to a Contract Manufacturing Organisation (CMO).
- The legal manufacturer assumes full responsibility for MDR compliance of the device, irrespective of who physically manufactures it.
- This legal manufacturer role under MDR is analogous to the MAH role in pharmaceutical regulation.
- The manufacturer is identified on the device labelling alongside the manufacturer symbol.
In the iDDC context, the MAH is responsible for the overall medicinal product but coordinates with device manufacturers to ensure the device component meets general safety and performance requirements (GSPR).
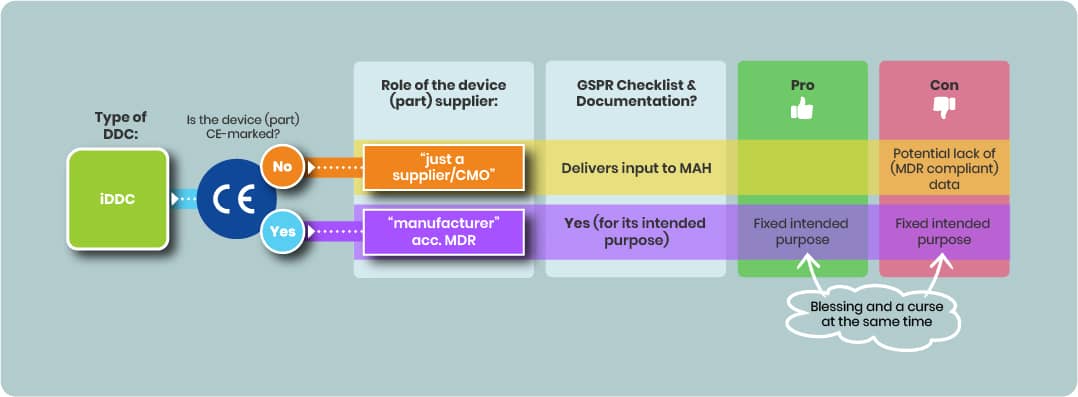
Using a CE-Marked Device Part
If a CE-marked device part is integrated:
- The iDDC’s regulatory status remains that of a medicinal product; the complete iDDC will not carry its own CE mark.
- Using a CE-marked device part may reduce compliance efforts with GSPR (see 2.1) if the device’s certified intended purpose aligns with the combined product’s intended purpose.
- However, the MAH must ensure that the intended use certification fits the actual use within the integral combination.
Using a non–CE-Marked Device Part
Selecting a non–CE-marked device component can be an alternative.
- Selecting a non-CE-marked device part offers flexibility to define or adapt the intended purpose but imposes significant regulatory challenges.
- However, the supplier of such a device part is considered only a supplier under MDR and assumes no legal responsibility for the device part. Thus there’s no GSPR checklist.
- The supplier of such a device part remains a supplier with no legal MDR responsibility for the iDDC’s device component, potentially complicating access to adequate regulatory data.
Importance of the Intended Purpose
The intended purpose is pivotal in medical device development:
- Determines device classification which defines the conformity assessment procedure needed for CE marking
- Defines the applicable GSPRs and
- Defines the design of the device and thus the manufacturing
Technical documentation must reflect verification and validation activities against relevant GSPRs, design, and production processes aligned with the intended purpose.
- For Class I devices, manufacturers may draw up an EU Declaration of Conformity without a Notified Body (NB).
- For devices above Class I, the TD and QMS must be submitted to an NB. Once certified, the manufacturer can issue the EU Declaration of Conformity.
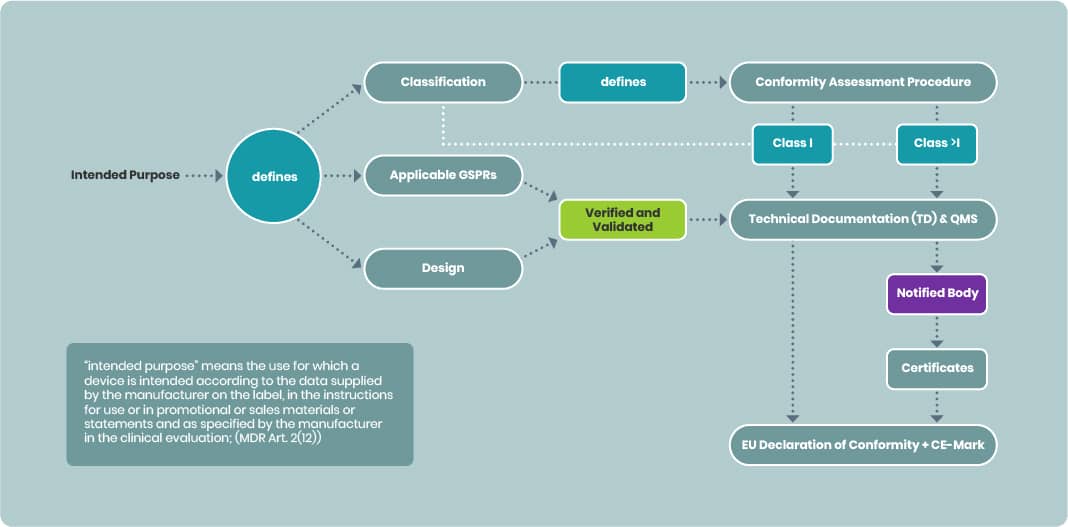
The signed EU Declaration confirms that the device is designed and manufactured to be suitable for its intended purpose during normal use (MDR Annex I).
Considerations for Co-packed Drug–Device Combination Products
Co‑packed drug–device combination products consist of a medicinal product and a medical device packaged together but remaining separate components. Unlike integral drug–device combinations (iDDCs), each component retains its own regulatory framework:
Medicinal product → EU pharmaceutical legislation, overseen by EMA or national authorities.
Medical device → EU Medical Device Regulation (MDR) 2017/745 requirements.
Regulatory Responsibility & Manufacturer Definition
The Marketing Authorisation Holder (MAH) of the medicinal product has overall responsibility for placing the co packed product on the market as a whole.
For the device component, the MDR (Art. 2(30)) defines a manufacturer as:
“A natural or legal person who manufactures or fully refurbishes a device, or has a device designed, manufactured or fully refurbished, and markets that device under their name or trademark.”
Key clarifications:
- The legal manufacturer is not necessarily the physical producer — manufacturing can be outsourced to a Contract Manufacturer (CMO).
- This legal manufacturer role under MDR is about regulatory responsibility, not production activity.
- The legal manufacturer must be clearly named on the device labelling, next to the manufacturer symbol.
In most co pack cases, the MAH is not the device manufacturer but may act as:
- A distributor under MDR; or
- An importer if sourcing the device from outside the EU; or
- The legal manufacturer if it chooses to assume that role (see Section 2.b).
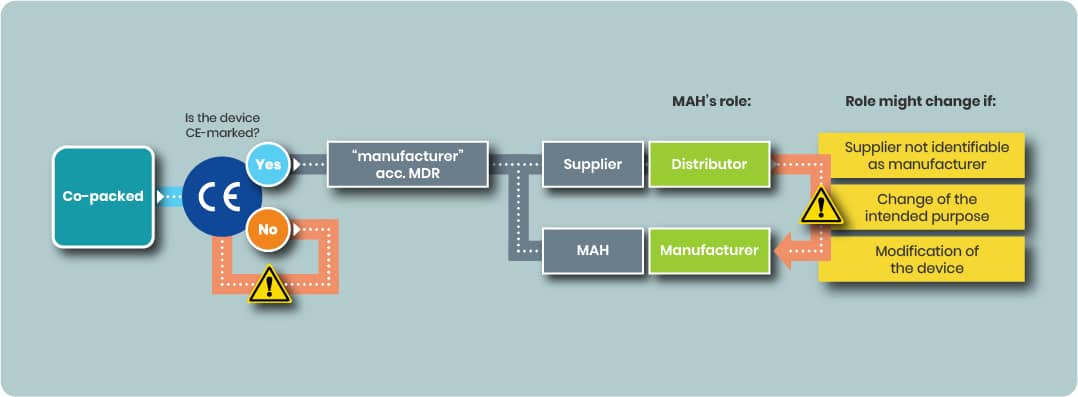
Using a CE Marked Device in a Co-pack
Case 1 MAH as Distributor
If the device part is already CE marked and:
- The CE mark remains valid,
- The intended purpose is unchanged,
- No modifications, relabelling, or repackaging affecting compliance are made, and
- The device manufacturer retains formal manufacturer obligations,
then, according to EMA guidance, the MAH’s obligations for the device part are those of a distributor (MDR Art. 14).
Distributor obligations include:
- Verifying CE marking, Declaration of Conformity, and required documentation.
- Ensuring the device is supplied with appropriate information and IFU in the correct language(s).
- Storing and transporting the device to maintain compliance.
- Maintaining traceability of the device (UDI requirements).
- Communicating incidents and recalls to the manufacturer and authorities.
- Cooperating with corrective actions.
Case 2 MAH as Legal Manufacturer
A MAH may choose to designate itself as the legal manufacturer for the device part.
- This means taking on full MDR manufacturer responsibilities under Art. 10.
- These include: drafting and maintaining technical documentation, ensuring GSPR compliance, implementing a QMS, undergoing conformity assessment, and meeting post market/vigilance requirements, etc.
- This route can be strategic — e.g. for supply chain control or centralised compliance management.
Using a Non CE Marked Device in a Co-pack
If the device lacks CE marking:
- It must undergo full MDR conformity assessment before using as in co-packed device.
- The MAH bears the compliance burden, including generating or obtaining full technical and clinical data, engaging a Notified Body (if applicable), and ensuring GSPR compliance.
- The supplier of such a part has no legal MDR responsibility for the co pack use, which can make data access challenging.
Importance of the Intended Purpose
The intended purpose determines refer to the iDDC section:
- The device classification;
- The conformity assessment route;
If the CE marked device’s certified intended purpose matches its function in the co pack, existing conformity can be maintained.
If not, the device may require reassessment and possible NB involvement before it can be marketed in the co pack.
Article 16(1) MDR — When Distributor Status Changes to Manufacturer
Under MDR Article 16(1), an importer, distributor, or other economic operator becomes the manufacturer — and assumes full Article 10 obligations — if they:
- Place the device on the market under their name/trademark without a clear contractual agreement of manufacturer responsibility to another party;
- Change the device’s intended purpose; or
- Modify the device in a way that affects MDR compliance.
Such changes trigger full manufacturer duties, including maintaining a compliant QMS, keeping and compiling & updating a technical documentation, and fulfilling post market surveillance and vigilance obligations.
Presumption of Manufacturer Status When the Original Manufacturer Cannot Be Identified
There is a presumption you are the manufacturer if the original legal manufacturer of the device is not clearly identified on the device or its packaging.
This situation is more likely when:
- The device is not separately packaged within the co pack; or
- The device has no stand alone Instructions for Use (IFU) distinct from the medicinal product’s documents, (for some class I or IIa devices)
In these cases:
- Device specific administrative information required under MDR must be provided separately, not embedded in the Patient Information Leaflet (PIL) or other medicinal product labelling.
- If this separate information is missing, the MAH may be presumed to be the manufacturer of the device component, with all associated MDR manufacturer obligations.
This presumption is explicitly addressed in the EMA FAQ on drug device combinations and can entail significant compliance consequences.
Documentation, Traceability & Post Market Responsibilities
- The device must have a UDI in accordance with MDR.
- The MAH is responsible for post market surveillance of the co packed product as a whole.
- Unless manufacturer status shifts under Art. 16(1), the device manufacturer retains its own MDR vigilance duties.
- The MAH’s MAA must include adequate information on the device to demonstrate safe and effective combined use.
Have a question or need more specific guidance?
We're more than happy to answer any of your inquiries:
CONTACT US TODAY